Мышечная дистрофия: причины, симптомы и лечение. Мышечная дистрофия у детей
Мышечная дистрофия — это группа патологий хронического наследственного характера, для которых свойственно прогрессирующее протекание, а также постоянные гистологические нарушения.
Современные методики исследования по части молекулярной генетики активно расширяют понимание и представление о большом количестве разновидностей дистрофии. Самые значимые из них — это мышечные формы дистрофии Беккера и Дюшена, а также состояния, передающиеся по наследству, по аутосомно-доминантному типу, прогрессирующая офтальмологическая форма мышечной дистрофии.
До настоящего момента не было изобретено средств, которые бы помогали полностью избавляться от дистрофии в мышцах.
Существует четыре формы данной патологии. Чаще всего ставится диагноз мышечной дистрофии Дюшена — в половине всех случаев патологии. Как правило. Течение болезни начинает уже в детском возрасте и провоцирует смертельный исход уже к двадцати годам. Мышечная дистрофия Беккера прогрессирует несколько медленнее, пациенты доживают до сорока лет. Другие формы заболевания обычно не оказывают никакого влияния на продолжительность человеческой жизни.
Этиологические факторы, вызывающие дистрофию в мышцах
Образование дистрофии в мышцах обусловлено влиянием разных генов. Патология Дюшена и Беккера вызвана генами, расположенными в половых хромосомах. Эти формы характерны только для представителей мужского пола. Другие поражения не соотносятся с половыми хромосомами, поэтому ими могут поражаться, как мужчины, так и женщины.
Основные проявления и признаки прогрессирования болезни
Все типы дистрофии в мышцах провоцируют активное развитие мышечной атрофии, но могут различаться в зависимости от степени тяжести патологии и времени её образования.
- Дистрофия Дюшена проявляется уже в раннем детском возрасте — примерно между тремя и пятью годами. Пациенты при этом ходят в развалку, трудно поднимаются по лестнице, часто подают на ровном месте и не могут бегать. Когда ребенок с таким диагнозом поднимает руки, то его лопатки будто отходят от туловища. Ребенок с этим типом дистрофии уже к 10 — 12 годам прикован к инвалидному креслу, а постоянно прогрессирующее ослабление мышц провоцирует смертельный исход от внезапно развившейся недостаточности сердца, недостаточности дыхательных процессов или инфекционных поражений.
- Дистрофия Беккера имеет большое количество общих черт с предшествующим типом патологии, но прогрессирует она намного медленнее. Симптомы мышечной дистрофии начинают проявляться только в возрасте пяти лет, а после пятнадцати лет пациенты ещё могут сохранять возможность ходить самостоятельно, порой даже и намного дольше.
- Плече-лопаточно-лицевая форма дистрофии мышц прогрессирует очень медленно, протекание у неё относительно доброкачественное. В основном болезнь дает о себе знать в возрасте 10 лет, но может проявляться и в начале подросткового периода. Дети с таким диагнозом уже в младенческом возрасте плохо сосут, а в более старшем возрасте у них не получается складывать губы в трубочку, приподнимать руки выше головы. Лицо отличается плохой мимикой во время плача или при смехе, но мимика временами всё же присутствует, несмотря на это, она сильно отличается от нормальной.
В центрах медицинской помощи, которые оборудованы самой современной техникой для реализации иммунологических и молекулярных обследований, специалисты могут точно установить тот факт, будет ли ребенок в будущем страдать от дистрофии в мышцах. В таких учреждениях также организуется обследования для родителей и для родственников ребенка и выявляется наличие у них генов, которые и определяют формирование мышечной дистрофии Беккера или Дюшена.
Как реализуется лечебный процесс?
В современной медицине ещё не было разработано методов предотвращения или прогрессирования активного развития данной патологии. Лечение мышечной дистрофии предполагает организацию противостояния осложнениям, например, позвоночным деформациям по причине слабых мышц спины, склонность организма к заражению воспалением легких из-за ослабления дыхательных мышц.
Пациентам с дополнительным развитием сердечной блокады может реализовываться имплантация водителя сердечного ритма. Для лечения сердечных поражений рекомендуется прием препарата фенигидина. Прием различных ортопедических позволяет укреплять висячие стопы, восстанавливать работу голеностопных суставов, а также уменьшить случаи падений.
Правильно подобранная тренировка тоже положительно влияет на протекание патологии. При развитии атрофии используются для лечения анаболические группы стероидов, а также общеукрепляющая терапия. При сильном проявлении миотонических симптомов поражения назначается курс лечения дифенином продолжительностью две — три недели. Именно дифенин предположительно угнетает патологическое воздействие на синаптическую проводимость, а также уменьшает посттетаническую мышечную активность. Препарат селегин дает положительные результаты при приеме его для коррекции режима сна и устранения высокой сонливости.
Эффективная терапия может проводиться только благодаря генному лечению, которое в настоящее время активно развивается. Большое количество экспериментальных работ свидетельствуют об улучшении состояния волокон мышц при терапии некоторых форм болезни. При развитии дистрофии Беккера и Дюшена происходит недостаточное производство белка мышц — дистрофина. Ген, который отвечает за образование данного белка — самый крупный ген из всех известных в медицине, в связи с чем, ученые воссоздали мини версию данного гена, а лучшими проводниками гена в мышцы стали аденовирусы.
В список прогрессирующих мышечных дистрофий (ПМД) входят миодистрофии:
- псевдогипертрофическая ;
- псевдогипертрофическая Беккера-Кинера;
- Эмери-Дрейфуса-Хогана;
- Роттауфа (фиброзирующая миопатия);
- ювенильная Эрба-Рота;
- окулярная (офтальмоплегия Грефе);
- плече-лопаточно-лицевая (Ландузи);
- окулофарингеальная;
- Дрейфуса;
- митохондриальные.
Причины мышечных дистрофий
У больных ПМД выявляется врожденный структурный дефект мышечной ткани (например, при ПМД Дюшенна — дефект гена, отвечающего за синтез структурного мышечного белка дистрофина). В отличие от утраты этого белка при миодистрофии Дюшенна, при миодистрофии Беккера дистрофин качественно изменен. У больных ПМД отмечают нарушения:
- возбудимости и проводимости мышечных волокон;
- микроциркуляции;
- нейротрофических влияний;
- метаболизма мышц.
Провоцирующими факторами выступают, в частности, инфекции, интоксикации, травмы (физические и/или психические) и соматические заболевания.
Симптомы мышечных дистрофий
Общие симптомы ПМД:
- мышечная слабость (симметричная);
- отсутствие перманентной боли;
- более частое проявление слабости в проксимальных отделах, ее преобладание в мышцах тазового, плечевого пояса;
- снижение и угасание сухожильных рефлексов пропорционально мышечной слабости.
В целом клиническая характеристика семейных и спорадических форм миопатий сходна. Прогрессируют медленно и постепенно. Локализация атрофий при различных формах миопатий:
- плечевые;
- тазовые;
- тазо-плечевые;
- плече-лопаточно-лицевые;
- дистальные;
- глазные;
- глазо-бульбарные;
- смешанные.
Характер распространения мышечной дистрофии — восходящий или нисходящий. Походка принимает так называемый утиный характер. Из положения лежа больные поднимаются с помощью дополнительных движений — приемов миопата. Наряду с атрофиями наблюдаются псевдогипертрофии мышц (у 37% больных), преимущественно в икроножных и четырехглавых мышцах, реже в дельтовидных, надостных, подостных и межреберных мышцах. Слабость мышц нарастают по мере развития заболевания и приводят к снижению мышечной силы, которую в поздних стадиях оценивают в 0-1 балл (по пятибалльной системе). Одновременно с нарастанием выраженности атрофий скелетных мышц отмечается понижение и угасание сухожильных надкостничных рефлексов. У большинства больных (83,5%) определяются вегетативно-сосудистые изменения: гипергидроз, акроцианоз стоп и ладоней, повышенная лабильность вазомоторов, стойкий красный дермографизм. Для различных форм первичных миодистрофий характерны и некоторые общие симптомы и феномены.
Миодистрофия псевдогипертрофическая Дюшенна
Псевдогипертрофическая мышечная дистрофия Дюшенна — злокачественная форма ПМД. Заболевание наследуется сцепленно с Х-хромосомой. При миодистрофии Дюшенна идентифицировано генетически обусловленное отсутствие структурного мышечного белка дистрофина, что ведет к запуску каскада химических реакций, вызывающих гибель миофибрилл. В соответствии с таким типом наследования болезни болеют обычно мальчики, матери которых являются носителями рецессивного гена. При миодистрофии Дюшенна существует эффект деда: от деда заболевание может передаться через дочь внуку.
Первые признаки мышечной дистрофии появляются на первом году, а к его концу становится заметным отставание детей в развитии. С 2 лет отмечается слабость мышц.
Основные проявления
«Утиная» походка
Пациент при ходьбе «переваливается» с ноги на ногу, что обусловлено слабостью ягодичных мышц
Псевдогипертрофии — «икры гнома»
Псевдогипертрофия икр из-за их жировой инфильтрации, разрастания соединительнотканных образований. Мышцы выпуклые, плотные на ощупь, но сила снижена (признак миопатии Дюшенна)
«Лягушачий живот»
При некоторых формах нервно-мышечной патологии низкий тонус мышц приводит при мышечной дистрофии к тому, что живот выступает вперед.
Симптом «вялых надплечий»
Если ребенка приподнять, взяв его подмышки, то предплечья резко поднимаются, и голова больного «тонет» между ними — признак выраженной гипотонии мышц плечевого пояса
«Поперечная» улыбка
Слабость и гипотрофия лицевой мускулатуры могут сопровождаться изменением мимики, в частности поперечным растягиванием рта. Признак некоторых форм миопатии.
Симптом Шерешевского-Говерса
Пациент с миопатией, вставая из положения лежа, совершает ряд движений (поворачивается на живот, становится на четвереньки), а затем начинает подниматься, постепенно разгибая ноги и опираясь руками; руки его последовательно меняют положение, при этом больной «вскарабкивается» по собственным ногам, как по приставной лестнице.
Триада синдромов при мышечной дистрофии: трех «А»
- Атрофии (гипотрофии)
- Атонии (гипотонии)
- Арефлексии (гипорефлексии) тазового пояса, вследствие чего возникает «утиная» походка.
Характерное распространение при мышечной дистрофии миодистрофического процесса восходящее — со временем в процесс вовлекаются все мышцы туловища и плечевого пояса. Грудная клетка уплощена, отмечают сколиоз грудного отдела и поясничный гиперлордоз. К 7 годам больные с трудом передвигаются, к 12-15 годам они утрачивают возможность ходить.
Характерны феномены Шерешевского-Говерса, поясничный гиперлордоз, псевдогипертрофия отдельных мышечных групп. Типична псевдогипертрофия икр («икры гнома»), при подъеме больные испытывают выраженные трудности. С вовлечением мышц плечевого пояса формируются «крыловидные» лопатки, кифосколиоз, развивается слабость рук, дыхательной мускулатуры.
В поздней стадии миодистрофического процесса возникают гипотрофия мышц лица, глотки и гортани; отмечается сгибательная контрактура в суставах конечностей. Развивается кардиомиопатия (расширение границ сердца, нарушения сердечного ритма), возникают изменения электрокардиограммы (ЭКГ), возможны адипозогенитальный синдром, гипоплазия надпочечников и остеопороз.
Около трети больных (30%) отстают в развитии интеллектуальных функций. В ранней стадии резко (в десятки и сотни раз) возрастает активность КФК, а также активность ЛДГ.
Миодистрофия псевдогипертрофическая Беккера-Кинера
Псевдогипертрофическую мышечную дистрофию Беккера-Кинера рассматривают как мягкую форму ПМД Дюшенна. Заболевание также передается сцепленно с Х-хромосомой. Дебют заболевания отмечается в возрасте от 5 лет.
Течение миодистрофического процесса медленно прогрессирующее. Особенности распространения мышечных дистрофий идентичны признакам мышечной патологии при ПМД Дюшенна. Менее выражена патология сердца. Интеллект пациентов сохранен; они долго сохраняют способность самостоятельно передвигаться, могут иметь детей.
Миодистрофия Эмери
Мышечная дистрофия Эмери-Хогана наследуется сцепленно с Х-хромосомой. Рано развиваются ретракции пяточных сухожилий, при ходьбе отмечают опору на пальцы, а также проявление «утиной» походки. Отмечают миокардиодистрофию, множественные контрактуры крупных суставов, ригидность позвоночника, бочкообразную грудную клетку. Активность КФК умеренно повышена. Течение медленно прогрессирующее. Интеллект сохранен.
Миодистрофия Роттауфа (фиброзирующая)
Дебют мышечной дистрофии происходит в детском возрасте, обычно в 4-12 лет. Возникают выраженные сухожильные контрактуры. Отмечают ограничения тыльного разгибания стоп, сгибания шеи. Вследствие фиброза мышц формируются патологические позы, приводящие к невозможности сгибания позвоночника. Миодистрофический процесс медленно прогрессирует. Появляются мышечная слабость и умеренные гипотрофии, больше в лопаточно-плечевой области. Интеллект сохранен. Развивается кардиомиопатия. Характерна выраженная гиперферментемия. ЭМГ выявляет изменения, указывающие на первичный миодистрофический процесс.
Ювенильная миодистрофия
В первую очередь болезнь характеризуется атрофией мышц таза. Ранним проявлением заболевания служит «утиная» (миопатическая) походка. Пациент испытывает затруднения при попытке сесть из положения лежа. Выявляют поясничный гиперлордоз, «лягушачий» живот, атрофии верхних конечностей (форма Лейдена-Мебиуса). Возможно развитие умеренных псевдогипертрофий. На фоне вовлечения в процесс межреберных мышц, диафрагмы может возникнуть дыхательная недостаточность.
У больных вероятны эндокринопатии (ожирение) и вегетативная дистония. Течение миодистрофического процесса относительно мягкое. При неблагоприятных условиях (например, при физических нагрузках) возможно быстрое прогрессирование процесса.
На ЭМГ при мышечной дистрофии присутствует картина первичного миодистрофического процесса. На ЭНМГ скорость импульса в пределах возрастной нормы. В крови определяется умеренная гиперферментемия.
Миодистрофия плече-лопаточно-лицевая (Ландузи)
Заболевание наследуется аутосомно-доминантно. Предположительно патологический ген локализован на хромосоме 4. Отмечается выраженная пенетрантность гена.
Дебют заболевания обычно происходит к 20 годам, болезнь начинается со слабости мышц лица. Рано наблюдаются «губы тапира», улыбка Джоконды). Со временем нарастает похудание и слабость передней зубчатой и большой грудной мышц. Позже миодистрофический процесс затрагивает перонеальную группу мышц, возникает походка «степпаж». Развивается умеренная псевдогипертрофия мышц. Креатин-креатининовый обмен нарушен умеренно.
Миодистрофия окулярная (Грефе)
Заболевание дебютирует до 30-летнего возраста. Нарастает поражение глазных мышц, которое протекает без диплопии, приводит к обездвиженности глазных яблок (параличу взора). Сначала часто поражается мышца, поднимающая верхнее веко; в поздней стадии миодистрофического процесса наблюдается двусторонний птоз. Иногда вовлекаются мимические, бульбарные и скелетные мышцы.
Миодистрофия окулофарингеальная
Окулофарингеальная миодистрофия — классическая наружная офтальмоплегия с дисфагией и дисфонией, наследуемая аутосомно-доминантно. В 1961 г. заболевание описано у представителей франко-канадской популяции.
Классический вариант болезни — прогрессирующая офтальмоплегия с дисфагией и дисфонией, сопровождаемая птозом верхних век. Другой вариант болезни протекает с присоединением пареза мышц, обеспечивающих движения глаз, мимической и жевательной мускулатуры, мышц шеи. При третьем, редком варианте в процесс вовлекаются также мышцы конечностей. Активность ферментов (КФК и ЛДГ) несколько повышена.
Миодистрофия Дрейфуса
Мышечная дистрофия Дрейфуса проявляется нарастающей слабостью мышц, преимущественно тазового пояса и нижних конечностей. Характерна походка больного с опорой на большие пальцы; отмечается поясничный гиперлордоз.
Особенность этой формы миодистрофии — формирование выраженных контрактур локтевых и других суставов. Нередко поражается миокард, больной отстает в психическом развитии.
Миопатии митохондриальные
При митохондриальных миопатиях биохимический дефект локализован в митохондриях клеток, что выявляют при биохимическом и ультрамикроскопическом исследованиях. Чаще всего дебют заболевания происходит на 2-м десятилетии жизни.
На ранних стадиях возникают птоз, наружный офтальмопарез (без диплопии вследствие симметричности поражения глазодвигательных мышц), слабость проксимальных мышцах, сухожильная гипо- и арефлексия.
Длительность прогрессирования мышечной дистрофии вариабельна: от месяцев до десятилетий. Клинико-нейрофизиологические исследования (ЭМГ, ЭНМГ) при митохондральных миопатиях выявляют миодистрофические и невропатические проявления.
Диагностика
Для постановки диагноза необходимы нейрофизиологические, биохимические и патогистологические исследования.
Локальная ЭМГ. У больных миопатиями регистрируется патологическая интерференция с высокой частотой полифазных потенциалов и укорочением времени формирования отдельных осцилляций.
Биохимическое исследование. На ранних стадиях активность КФК крови увеличивается в 50 раз и более; ЛДГ — в 5-7 раз; ФДА — в 2-5 раз; в поздних стадиях снижается до возрастной нормы.
Патогистологическое исследование. Определяются признаки ПМД:
- перерождение мышечной ткани;
- беспорядочное расположение разнокалиберных мышечных волокон;
- чередование нормальных, атрофированных и (в некоторых мышцах) гипертрофированных волокон;
- атрофия мышечных волокон в длину и в поперечнике;
- пролиферация ядер с их расположением под сарколеммой и внутри волокна.
Лечение мышечных дистрофий
Лечение первичных направлено на замедление темпа развития заболевания и максимальное сохранение способности больного к самообслуживанию.
Тактика лечения ПМД:
- диетотерапия с целью избежать развития у больного избыточной массы тела;
- и двигательная активность, в частности, для поддержания функций опорных суставов и предотвращения контрактур;
- дыхательная гимнастика;
- медикаментозная терапия;
- социально-психологическая реабилитация;
- ортопедическая помощь.
При некоторых формах миодистрофий, в частности при миодистрофии Дюшенна, применение глюкокортикоидов иногда позволяет отодвигать наступление обездвиженности больного на годы. Поскольку такое лечение длительное и обычно сопровождается многими осложнениями, необходимо обдуманное назначение глюкокортикоидов. При ПМД применяют (с учетом возможных противопоказаний) преднизолон 1-2 мг/кг утром, через день, возможен и ежедневный прием преднизолона при мышечной дистрофии (утром однократно в дозе 0,75 мг/кг/сутки). Авторы предлагают применять также препарат этой же группы — дефлазакорт, обладающей меньшими побочными эффектами, приблизительно в той же дозе (по эффективности 6 мг дефлазакорта соответствуют 5 мг преднизолона). Длительность лечения зависит от эффективности и выраженности побочных действий препарата.
Показана симптоматическая и общеукрепляющая терапия. В частности, рекомендуют применение аденозина фосфата, трифосаденина, витамина Е, коферментов (например, кокарбоксилазы), негормональных анаболических средств (этилтиобензимидазола, оротовой кислоты), инозина, препаратов калия. При этом надо иметь в виду недоказанность объективных проявлений эффективности этих препаратов.
Преднизолон внутрь (утром) по 1-2 мг/кг/сутки через день, длительность терапии определяют индивидуально, или Преднизолон внутрь (утром) по 0,75 мг/кг/сутки ежедневно, длительно или Дефлазакорт внутрь по 6 мг, длительно.
Прогноз при мышечных дистрофиях
Прогноз наследственных миопатий зависит от формы. Поскольку у большинства больных заболевание носит неуклонно прогрессирующий характер, то прогноз неблагоприятный.
Больные миодистрофией псевдогипертрофической Дюшенна обычно умирают на 3-м десятилетии жизни, чаще от прогрессирующей сердечной недостаточности. При псевдогипертрофической мышечной дистрофии Беккера-Кинера и мышечной дистрофии Эмери-Дрейфуса-Хогана больные обычно доживают до 40-60 лет. При миодистрофии Роттауфа-Мортъе-Бейера пациенты нередко доживают до 30-50 лет.
Статью подготовил и отредактировал: врач-хирургМышечная дистрофия входит в группу заболеваний, вызывающих дегенерацию скелетных мышц и суставов. Препаратов для его лечения пока не существует, хотя есть эффективные медицинские процедуры, способные замедлить развитие заболевания. Симптомы могут быть слабо выраженными, но в тяжелых случаях болезнь ухудшает рефлексы и лишает больного способности ходить.
Разновидности мышечной дистрофии
Выявлено более 30 видов мышечной дистрофии, из них наиболее распространены 9:
- миодистрофия Дюшенна – наиболее распространенная форма, развивается преимущественно у молодых мужчин;
- дистрофия Беккера – к сорокалетию больной теряет способность самостоятельно передвигаться;
- миотония – симптомы проявляются чаще у взрослых (либо у детей в период позднего детства);
- врожденная дистрофия – диагностируется сразу после рождения или в первые месяцы жизни;
- лимб-поясная – свойственна подросткам, молодым людям (20-25 лет), затрагивает мышцы бедер и плеч;
- плече-лопаточно-лицевая – вызывает ослабление мышц лица, плечевых мышц, теряется способность поднимать руки, возникают проблемы с речью, тахикардия;
- дистальная – для заболевания характерна слабость рук и нижних конечностей;
- дистрофия Эмери-Дрейфуса – для женщин опасна параличом сердца, у детей (преимущественно мальчиков), кроме сердца, поражаются икроножные мышцы и верхний плечевой пояс;
- болезнь Шарко (и прочие дегенеративные заболевания) также могут провоцировать схожие с мышечной дистрофией симптомы.
Из этих 9 форм мышечной дистрофии наиболее часто диагностируют следующие:
- Дистрофия Дюшенна. Симптомы проявляются в раннем детстве (до 5 лет). Болезнь диагностируют у одного из 3500 новорожденных мальчиков. Это наиболее распространенное смертельное генетическое расстройство. Женщины могут быть носителями гена, подвергшегося мутации, но сами этим расстройством не страдают. Дегенеративные изменения блокируют белок (дистрофин), поддерживающий структурные элементы мышечной ткани и клеточных мембран. Без дистрофина мышечная слабость прогрессирует, провоцируя обездвиженность и смерть.
- Дистрофия Беккера. Хотя производство дистрофина при этом заболевании сохраняется, уровень стержневидного белка существенно ниже нормы. Мышечная дистрофия развивается к 12-летнему возрасту. Болезнь развивается медленнее, но поражения значительны: искривление позвоночника, трудности с дыханием, постоянная усталость, слабость, болезни сердца, когнитивные проблемы.
Основные симптомы дистрофии
Хотя все расстройства связаны между собой, каждый тип дистрофии вызван уникальной мутацией генов, поэтому симптомы и время проявления заболевания разнятся:
- потеря мышечной массы (проблема усугубляется с возрастом);
- поражение нижних конечностей (далее слабость распространяется на мышцы шеи, плечи, спину, грудь);
- прогрессирующее истощение;
- увеличение икроножных и дельтовидных мышц;
- снижение гибкости, выносливости, усталость;
- нарушение координации, синхронизации движений;
- падения (из-за потери координации и мышечной слабости);
- скованность суставов, боль при движении;
- замена фиброзной ткани мышечной;
- трудно приседать, наклоняться, подниматься по лестнице;
- аномальное развитие скелета (кривизна позвоночника, нарушение осанки);
- уменьшается диапазон движения (до полного паралича);
- развивается пневмония, другие проблемы с работой дыхательной системы, нарушается сердечная деятельность, что может привести к смерти (при дистрофии Дюшенна).
Дистрофия передается по наследству. Однако в 35% случаев заболевание провоцирует спонтанная мутация генов из-за нарушения выработки белка. Мужчины подвержены большему риску из-за наличия одной Х-хромосомы. Y-хромосома (ещё одна половая хромосома, имеющаяся у мужчин) не имеет копии гена дистрофина и на развитие мышечной дистрофии влияния не оказывает.

У женщин риск дистрофии уменьшается благодаря наличию двух Х-хромосом. Даже если одна из хромосом аномальна, здоровый ген может производить достаточно дистрофина. Вероятность передачи по наследству дефектного гена составляет 50%.
Диагностика мышечной дистрофии
При диагностике дистрофии проводят:
- физический экзамен – проверяется гибкость, тестируется мышечная сила, диапазон движения и другие характеристики пациента;
- электромиографию – исследуется электрическая активность внутри мышц;
- электрокардиограмму;
- биопсию мышц – позволяет установить, какой тип дистрофии развивается;
- магнитно-резонансную томографию – показывает, какие органы и ткани поражены.
Женщины могут пройти тест при планировании беременности, чтобы знать о наличии или отсутствии дефектного гена. По анализу крови можно определить наличие ферментов, связанных с аномальным развитием мышц.
Лечение мышечной дистрофии
Лечение зависит от типа дистрофии. Дистрофия Беккера и Дюшенна неизлечимы, однако есть средства, позволяющие управлять симптомами болезни. Чем больше методик комбинируется (включая диету, эмоциональную и физиотерапию, лекарственную поддержку), тем лучше результат.
Терапия может включать:
- прием стероидов (для борьбы с мышечной слабостью и снятия болевых симптомов), альбутерола (при наличии астмы), лекарств, улучшающих работу сердца (ангиотензина, бета-блокаторов), ингибиторов протонной помпы, биодобавок, позволяющих поддерживать энергию и мышечную массу;
- использование ортопедических изделий (скобы, инвалидные кресла);
- лечение патологии речи (если затронуты лицо и язык).

Естественные методы управления мышечной дистрофией
Физическая активность
Важно пытаться сохранить гибкость и силу мышц. Пассивный режим, отсутствие элементарных нагрузок усугубляют симптомы болезни, способствуют развитию осложнений и эмоционального расстройства. Для поддержания гибкости и координации тела полезна физиотерапия. Мобильность можно повысить, используя ортопедические вкладки, корсет, трость, электроскутер, коляску, можно двигаться при поддержке ассистента.
От боли в суставах, для улучшения балансировки, уменьшения тревожности, сохранения диапазона движений полезно плавание, йога, упражнения на растяжку (для страдающих мышечной дистрофией разработаны специальные практики, найти их можно в интернете или у врача).
Психологическая поддержка
Здоровая диета
Некоторые пищевые химические вещества воздействуют на геном человека и меняют структуру генов, провоцируя возникновение или прогрессирование хронических заболеваний, развитие осложнений. Воспалительные процессы поддерживает обедненная полезными веществами (или перегруженная обработанными продуктами) диета, отсутствие физической активности, стресс. Эти факторы снижают способность организма защищать клетки от дегенерации, ускоряют старение, приводят к потере плотности костной ткани.
Противовоспалительная диета хоть и не сможет исцелить от мышечной дистрофии, но существенно замедлит прогрессирование болезни. Такой рацион уменьшает воспаления, подщелачивает организм, снижает уровень глюкозы, помогает вывести токсины, обеспечивает питательными веществами.
Принципы питания:
- замена «плохих» жиров «хорошими» – отказ от гидрогенизированного масла (соевого, рапсового), трансжиров, введение полезных насыщенных и ненасыщенных жиров (оливкового, подсолнечного, кокосового, льняного, кунжутного масла, авокадо, омега-3 жиров);
- выбор органических мясопродуктов (без антибиотиков, гормонов);
- исключение рафинированного сахара и глютена.
Лучшие противовоспалительные продукты — листовые овощи, китайская капуста, сельдерей, свекла, брокколи, черника, ананас, лосось (дикий), костный бульон, грецкие орехи, куркума, имбирь, семена чиа.

Кроме чистой воды, полезны травяные чаи, свежевыжатые соки, натуральный лимонад, морс, квас. Из молочных продуктов допустимо оставить в меню козье молоко (сыр, йогурт из него), овечий сыр.
Важно исключить вероятность попадания в организм токсинов не только из пищи, но и из бытовой химии, косметики, выбирая натуральные средства.
Добавки, поддерживающие мышечную ткань
По назначению врача, при мышечной дистрофии можно использовать следующие биодобавки:
- аминокислоты (карнитин, коэнзим Q10, креатинин) – способствуют выработке белка, необходимого для поддержки мышц;
- глюкозамин и хондроитин – борются с суставной болью;
- антиоксиданты (витамины С, Е, А) – полезны для сердца, суставов и мышц;
- зеленый, матча чай (экстракты) – поддерживают энергетический уровень;
- омега-3 кислоты, рыбий жир – противостоят воспалениям;
- пребиотики – улучшают пищеварение.
Эфирные масла
Уменьшить отек, снять боль и другие симптомы, вызванные дегенерацией мышц, тканей и суставов, помогает масло мяты, ладана, имбиря, куркумы, мирры. Эффективно при лечении депрессии, для снятия тревожности масло лаванды, ромашки, грейпфрута, сандалового дерева. Можно добавлять масло по капле в диффузор увлажнителя воздуха, принимать на его основе ванны и использовать для массажа.
При обнаружении любых тревожных признаков (онемении лица, нарушении речи, походки, внезапной слабости, снижении гибкости) необходимо пройти обследование. Раннее вмешательство позволит затормозить развитие болезни и уменьшить негативное влияние симптомов мышечной дистрофии на качество жизни.
В современной неврологии существует огромное количество заболеваний, характер возникновений которых не поддаются рациональному объяснению со стороны специалистов. К таким болезням можно отнести группу таких недугов, как мышечная дистрофия. Всего разновидностей данной болезни девять, но обо всем по порядку…
Мышечная дистрофия - это хроническое наследственное заболевание в результате развития которого происходит поражение мышечной системы человека. Пораженные мышцы перестают нормально функционировать, истончаются в размерах, а в месте их расположения в организме постепенно нарастает жировая прослойка.
Разновидности мышечной дистрофии
В современной неврологии данный недуг классифицируется на девять различных болезней. Классификация заболевания связана с:
- локализацией мышечных нарушений;
- характеристиками болезни;
- агрессивностью развития;
- возрастом.
Так, мышечная дистрофия бывает:
- Дюшенна;
- миотоническая (болезнь Штейнерта);
- Беккера;
- Эрба Рота;
- юношеская форма дистрофии Эрба-Рота;
- плече - лопаточная лицевая форма (Ландузи-Дежерина);
- алкогольная миопатия;
- дистальная форма;
- миодистрофия Эмери -Дрейфуса.
Дистрофия Дюшенна
Наиболее известная форма прогрессирующая дистрофия Дюшенна (псевдогипертрофическая дистрофия, мерозин - негативная врожденная дистрофия). Данная разновидность недуга проявляется в детском возрасте начиная с двух до пяти лет. В первую очередь от данного недуга страдают мышцы нижних конечностей, которые несмотря на малоподвижный образ жизни у маленьких пациентов, постепенно увеличиваются в размерах. Данная особенность связана с нарастанием жировой ткани вместо мышц.

Слева здоровый ребенок, справа больной
Постепенно, по мере прогрессирования болезни она переходит в верхнюю часть и поражает мышцы верхних конечностей. Как правило, уже к двенадцатилетнему возрасту маленький пациент перестает полностью двигаться. Летальность у данного заболевания очень высока примерно 85–90% больных не доживают до 20-летнего возраста.
В группе риска находятся представители мужского пола, так как девочек данный недуг практически не затрагивает.
Болезнь Штейнерта
Врожденная дистрофия Штейнера не зря имеет название миотоническая, так как в результате прогрессирования болезни происходит слишком медленное расслабление мышц после их сокращения (такое явление называется миотонией).
Данное заболевание, в отличие от предыдущего распространено у взрослых, в возрасте от 20 до 40 лет. Имеют место быть и случаи прогрессирования болезни у детей, как правило, в младенческом возрасте, однако, это скорее исключение, чем правило.

На лицо признаки миотонии (приоткрытый рот и веки)
Гендерной зависимости болезнь не имеет и в равной степени от нее страдают как мужчины, так и женщины. Отмечается, что при недуге проявляется слабость мимических мышц лица, а также конечностей. Прогрессирование, в отличие от болезни Дюшенна происходит медленно.
Отличительная черта заболевания, возможность поражения не только мышц конечностей, но и внутренних мышц (сердечная мышца), что в свою очередь, представляет большую опасностью для жизни человека.
Болезнь Беккера
Данная форма болезни также долго прогрессирует, и пациент длительное время чувствует себя хорошо. Обострение недуга может произойти на фоне полученных травм или различных заболеваний нервной системы, которые своим течением ускорят развитие недуга.
В группе риска находятся люди низкого роста.
Болезнь Эрба-Рота и юношеская ее форма
Данное аутосомно - рецессивное заболевание развивается у пациентов после 20 лет, а юношеская форма у детей и подростков 11-13-летнего возраста. Прогрессирование болезни происходит по восходящему варианту, то есть сначала поражаются мышцы нижних конечностей, и постепенно происходит восхождение недуга к верхним конечностям.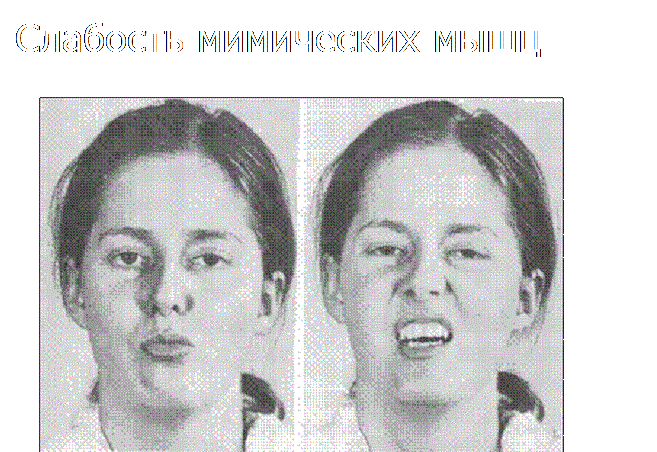
Отличительной чертой недуга является наличие выступающих лопаток, которые по мере прогрессирования выступают более явно и очевидно.
Во время ходьбы отмечается переваливание пациента, выпячивание живота и задвигание грудной клетки.
Болезнь Ландузи-Дежерина
Плече-лопаточно-лицевая форма данного заболевания является наиболее безразборным видом, так как поражает людей в возрасте от пяти до 55 лет. Для данного недуга характерно очень длительное развитие, пациент может сохранять трудоспособность до 25 лет жизни с данной болезнью.
Отличительными симптомами является поражение лицевых мышц, в результате чего у больного могут возникнуть проблемы с четкостью произношения, в связи с неполным смыканием губ. Кроме того, отмечается неполное смыкание век.
По мере развития у больного атрофируются лицевые мышцы, мышцы плеч, конечности и туловища.
Данная форма не зависит от генетической мутации.
Алкогольная миопатия
Данный вид болезни также несвязан с генными мутациями и причина его возникновения одна - злоупотребление алкоголем. У больных могут отмечаться болевые синдромы в конечностях, связанные именно с поражением мышц.
Различают острую и хроническую алкогольную миопатию.
Дистальная форма
Дистальная форма мышечной дистрофии является доброкачественным вариантом прогрессирующей дистрофии. Как правило, данное заболевание трудно дифференцировать от невральной амиатрофии Мари-Шарко. Для разделения этих двух заболеваний требуется проведение электроэнцефалограмму головы, которая и дает понимание с какой болезнью предстоит иметь дело.
К основным симптомам недуга относят атрофию мышц конечностей с их последующим исхуданием. Возможны парезы стоп, кистей рук и т. п.
Миодистрофия Эмери -Дрейфуса
Данный тип болезни первоначально, вообще, не был выделен в качестве отдельного заболевания, так как по своей симптоматике был схож с дистрофией Дюшенна. Однако, в дальнейшем в результате длительного исследования было установлено, что болезнь Эмери -Дрейфуса обладает индивидуальной симптоматикой.
Болезнь относят в разряд редких. В группе риска находятся люди до 30 лет, как правило, юный возраст. Есть свидетельства о проявлении симптомов и после 30 лет, но они единичны.
Основное отличие данного вида болезни - это проблемы с мышцами сердца, которые в итоге могут стать причиной смерти. Кардиомиопатия при данном недуге не единственное отличие. Кроме проблем с сердцем, у пациентов наблюдают стандартные для дистрофии Дюшенна признаки, но в более щадящем режиме развития.
Причины
Негативная составляющая болезней нервной системы заключается в том, что они тяжело поддаются изучению. По этой причине не до конца известно о факторах, провоцирующих развитие той или иной формы мышечной дистрофии.
Основная причина, для формирования большинства подвидов данной болезни является генная мутация, в частности гена, который отвечает за синтез белка.
К примеру, болезнь Дюшенна имеет прямое отношение к мутации половой Х хромосомы. Основной переносчик нехорошего гена выступают девушки, которые несмотря на его наличие в собственной ДНК не страдают от данной болезни.
Что касается миотонической формы, то виновник ее возникновения ген, расположенный в 19 хромосоме.
Основные симптомы
Наличие большого количество различных подвидов у данного недуга указывает на различия в симптоматике, однако у болезни есть общие симптомы, которые в себя включают:

Диагностика мышечной дистрофии
Диагностические мероприятия при подобном недуге обширны, так как существует большое количество болезней, от которых необходимо дифференцировать болезнь.
На первоначальном этапе доктор обязательно изучит анамнез больного, уточнит о симптоматике, режиме дня и т. п. Эти данные требуются для составления плана проведения последующих диагностических мер, которые могут в себя включать:

Стоит отметить, что чем позже проявилась болезнь, тем лучше для больного, так как ранние симптомы в большинстве случаев заканчиваются смертью.
Лечение
Лечение мышечной дистрофии процесс сложный и затяжной, однако, в настоящее время не создано лекарство, которое полностью исцеляет больного. Все мероприятия направлены на облегчение жизни больного и восстановления некоторых утраченных способностей.
Для затормаживания развития болезни назначают следующие препараты:
- кортикостероиды;
- витамины В1;
- аденозинтрифосфат (АТФ).
Кроме того, для замедления процесса развития используют фетальные стволовые клетки, которые замедляют процесс дистрофии.
Помимо того, в качестве профилактических мероприятий назначают:
- массаж;
- физиотерапию;
- дыхательную гимнастику.
Помимо стандартных вариантов лечения, важно постоянно руководствоваться тремя главными составляющими в процессе жизнедеятельности:
- Адекватная физическая активность.
- Своевременная психологическая поддержка.
- Соблюдение диеты.
Физическая активность
Отсутствие желания у человека бороться с недугом оказывает негативное воздействие на организм. Судите сами, пассивность, нежелание двигаться угнетает и без того пораженную мышечную систему. Мышцам необходимо давать нагрузку, так как без нагрузки дистрофичные процессы начинают происходить быстрее, тем самым прогрессируя в более быстром темпе.
Умеренная физическая активность, использование поддерживающих приспособлений будут отличным подспорьем в борьбе с недугом.
При наличии болей в мышцах отлично подойдет плавание, йога, упражнения на растяжку.
Психологическая поддержка
Психологическая поддержка окружения важна для больного человека. А если недуг такой серьезный, как этот тем более. Для кого-то будет достаточно обычной поддержки со стороны друзей и родных, а кому-то может потребоваться квалифицированная психологическая помощь.
Важно дать понять такому человеку, что он не остался один на один со своей проблемой. Он должен понимать, что ему есть к кому обратиться, есть люди, которые сопереживают и поддерживают его.
Диета
Что касается режима питания и соблюдения диеты, существует расхожее мнение, что соблюдение противовоспалительной диеты может замедлить прогрессирование недуга. Данная диета снижает воспаление, уровень глюкозы, выводит токсины из организма и питает его полезными веществами.
Суть такой диеты в следующем:
- Отказ от продуктов, содержащих «плохие» жиры и замена их на «хорошие», введение в рацион ненасыщенных жиров, которые содержатся в оливковом, льняном, кунжутном масле, авокадо.
- Применение в пищу мяса и рыбы, при производстве которых не были использованы антибиотики или гормоны.
- Полное удаление из рациона рафинированного сахара и глютена.
- Употребление в пищу следующих продуктов - китайская капуста, брокколи, сельдерей, ананас, лосось, свекла, кукумария, имбирь, куркума и других продуктов, обладающих противовоспалительными свойствами.
- Молочные продукты допустимы только на основе овечьего и козьего молока.
- Допустимо употребление травяных чаев, лимонада, кваса, морсов и натуральных соков.
Профилактика
Так как мышечную дистрофию довольно тяжело предсказать и обнаружить на раннем этапе, профилактические мероприятия сводятся к двум простым рекомендациям:
Обязательное обследование женщины на этапе планирования беременности на предмет наличия мутаций в генах
В случае если до беременности, по каким-либо причинам не было сделано генетического обследования, уже в период беременности проводится тест на определение у плода мутаций в Х хромосоме.
Прогноз и осложнения
В зависимости от вида болезни прогноз может быть разным, и тем не менее можно выделить несколько возможных осложнений, возникающих при данном недуге.
- нарушения сердечной деятельности
- искривление позвоночника
- снижение интеллектуальных способностей больного
- снижение двигательной активности
- нарушения дыхательной системы
- летальный итог
Итак, мышечная дистрофия довольно опасное и неизлечимое заболевание, поэтому будущим родителям необходимо с глубокой ответственностью относиться к планированию беременности. Берегите себя и своих будущих детей!
Дело в том, что я заглянул в электронный микроскоп и увидел (изображен белым цветом на изображении) между мышечными волокнами (красный цвет).
На фото : биопсия мышечных волокон при легкой (A), средней (B) и тяжелой степени миопатии (C):
На фото : нормальные мышечные волокна здорового человека:

На примере моей пациентки, которая страдала . Диагноз Эмине: мышечная дистрофия тяжелой степени тяжести , подтвержденная биопсией. Дальше я опишу свой подход к устранению слабости в мышцах. Рекомендую посмотреть видео по теме лечения прогрессирующей мышечной дистрофии Дюшенна.
Мышечная дистрофия - это болезнь нарушения создания белка, создающего каркас мышечной клетки.
- Образуются дыры в каркасе клетки. Из этих дыр вытекают жизненно важные соединения и микроэлементы. Чтобы залатать дыры, клетка вынуждена вырабатывать вещества, которые больше этих дыр. Клетка изнутри «раздувается», т.е. отекает.
- Увеличивающийся отек давит на мышечные клетки снаружи, отодвигая ядра клеток и митохондрии к периферии.
- В клетке повышается уровень креатинфосфокиназы и мышца теряет способность связывать и удерживать креатин.
- Креатин нужен митохондрии для выработки энергии в мышечной клетке.
- Митохондрии уменьшают вырабатывать АТФ. АТФ - это энергия, необходимая для работы двигательных белков актина и миозина. Нет энергии - нет движения.
- Внутри мышечного волокна, которое находится без движения, замедляются свои собственные процессы питания.
- Мембрана волокна начинает выделять ненужные ей без функции движения ферменты и аминокислоты. Поэтому возникла теория “дефектных мембран”.
- Во время движения мышцы эти ферменты и аминокислоты нужны. Для их синтеза нужна энергия, которой нет. Поэтому возникает мышечная слабость.
- Начинается атрофия мышечных волокон.
Симптомы
Заболевание мышечная дистрофия начинается с развития слабости и атрофии определенной группы мышц. С годами дистрофический процесс захватывает все больше новых групп мышц. Это происходит вплоть до полной обездвиженности. Основной признак миодистрофии - это поражение мышц тазового, плечевого пояса и туловища больного. Мышцы бедра и мышцы плеча поражаются в тяжелых случаях, что и было у пациентки Эмине: она не могла вставать без опоры и ходить, даже, небольшие расстояния.
Мышечные дистрофии двусторонние
В начальный период может преобладать миодистрофия с одной стороны, но по мере развития заболевания степень поражения становится одинаковой в симметричных мышцах больного. Со временем течения болезни практически во всех мышцах снижается их мышечная сила. На теле больного, страдающего мышечной дистрофией, появляются участки гипертрофических мышц. Это псевдогипертрофия, которая не связана с увеличением мышечных волокон. Псевдогипертрофия мышц связана с отеком в мышцах ног или рук. Такие мышцы плотные, но слабые.
Формы мышечной дистрофии у взрослых
Все образные формы рассматриваемого заболевания у взрослых отличаются:
- типами наследования;
- быстротой и характером его течения;
- наличием или отсутствием сухожильных ретракций и псевдогипертрофий;
- сроками начала процесса;
- своеобразием топографии мышечных страданий;
- другими признаками прогрессирующей мышечной дистрофии.
Вопросы классификации миопатии (хронические и прогрессирующие наследственные заболевания мышц) разрабатываются в разных направлениях. Мышечную дистрофию у взрослых классифицируют по типу наследования:
- Аутосомно-доминантные.
- Аутосомно-рецессивные.
- Доминантные и рецессивные.
- Сцепленные с Х-хромосомой.
Обследование при миопатии
Клиническими признаками, характерными для мышечной дистрофии, являются симптомы вялого паралича в разных группах мышц больного человека без признаков поражения двигательных нейронов и периферических нервов. Неврологи всего мира не могут это объяснить.
Доктор Никонов
Мое мнение: белковый отек между мышечными волокнами делает невозможным движения мышц.
Не знание этого феномена приводит в недоумение врачей всего мира: “Как так? Мышечное волокно целое, не поврежденное. Двигательные нейроны и периферические нервы целые, находятся на своих местах и отлично пропускают импульсы, идущие от головного мозга к мышцам и от мышц к головному мозгу, а движения затруднены?”.
Неврологи назначают сделать электромиографию. И опять для них загадка: нет изменений в структуре мышечного волокна. Снижение амплитуды М-ответа, усиление интерференции и полифазности потенциала говорит о затруднении в движении мышц без какой-либо патологии!
Патологоанатомическая картина заболевания
Посмотрим, что происходит внутри мышечных клеток у пациентов, страдающих миодистрофией Дюшенна. Для этого сделаем надрез кожи, расширим расширителем и возьмем небольшой кусочек мышечных волокон.


Типичный признак миодистрофии в первую очередь - это разный диаметр мышечных волокон. У здорового человека диаметр мышечных волокон одинаковый.
Характерные признаки мышечной дистрофии - атрофированные и гипертрофированные волокна, множественные внутренние ядра и отек.
Изучая окрашенные срезы скелетной мышцы, я увидел денервацию миофибрилл, значительный разброс в размере миофибрилов и выраженный отек.

Пояснение к первому фото :
- Бледно-фиолетового цвета - это мышечные волокна в разрезе.
- Светлые пятна как внутри волокон, так и снаружи - это отек.
- Темные точки - это ядра, которые отек сместил к периферии.
На втором фото показано нормальное мышечное волокно здорового человека.
Степень тяжести мышечной дистрофии по данным электронной микроскопии ориентируется на следующие показатели:
- при легкой степени разница в размере мышечных волокон выражена умерено, начальные признаки отека (белый цвет).
На фото : биопсия мышечных волокон при легкой (A), средней (B) и тяжелой степени дистрофии (C).
- средняя степень тяжести соответствует перемещению ядер в центр мышечных волокон, расширению межфибриллярного пространства из-за увеличения отека между клетками.
На фото: мышечные волокна при прогрессирующей мышечной дистрофии средней степени тяжести:
а) светло-фиолетового цвета мышечные волокна;
б) светлые пятна внутри мышечных волокон - отек, оттолкнувший ядра из центра клетки к периферии;
в) темные точки - ядра мышечных клеток;
г) стрелкой показана мышечная клетка, которая не может двигаться из-за уменьшения обменных процессов, - темнеет в сторону фиолетового цвета.

- тяжелая степень характеризуется обширными очагами деструкции миофибрилл, их фрагментация и дезорганизация, появление гиалиноподобного вещества и отека между мышечными клетками. Функционально такая ткань обладает слабой силой, быстро наступает утомление и развиваются признаки мышечной усталости. Фото будет представлено немного ниже.
Вот такое состояние мышц было у Эмине до обращения ко мне:

Пояснение к фото “тяжелая степень мышечной дистрофии”:
- Мышечные волокна в разрезе окрашены в синий цвет.
- Красные точки - это ядра мышечных клеток.
- Отек - неокрашенный белый цвет.
Клиническая картина мышечной дистрофии
Первым признаком миопатии Дюшенна у Эмине была слабость. Она стала утомляться при обычных физических нагрузках. Наиболее ранними жалобами у Эмине были:
- Утомление при беге, длительной ходьбе.
- Эмине стала часто падать.
- Стали появляться миалгии в ногах (боль в области мышц), иногда в комбинации с болевыми спазмами.
- Постепенно ходьба стала затрудняться.
Встать из низкого стула Эмине не могла без помощи рук. При вставании женщина прибегала к использованию вспомогательных приемов: “вставание лесенкой», «взбирание по самому себе” - прием Говерса. Еще через несколько лет Эмине не могла встать с корточек без помощи. Пациентка не могла подниматься по лестнице.
После моего воздействия на мышцы Эмине, она поднимается на 17 этаж без помощи рук, сразу же спускается на лифте вниз и снова поднимается на 17 этаж, не уставая!
Атрофия мышц развивается преимущественно в области тазового пояса, бедер (поэтому эмендическое воздействие на мышцы Эмине было направлено на эти участки).
Мышцы верхних конечностей начинают атрофироваться позже. Эмине говорила, что не может налить себе чай или расчесаться. Посмотрите на результаты лечения мышечной дистрофии на представленном ниже видео: