Distrofia musculara: cauze, simptome si tratament. Distrofia musculara la copii
Distrofia musculară este un grup de patologii de natură ereditară cronică, care se caracterizează printr-un curs progresiv, precum și prin tulburări histologice permanente.
Metodele moderne de cercetare în ceea ce privește genetica moleculară extind în mod activ înțelegerea și înțelegerea unui număr mare de varietăți de distrofie. Cele mai semnificative dintre ele sunt formele musculare ale distrofiei Becker și Duchenne, precum și afecțiunile care sunt moștenite, după un tip autosomal dominant, o formă oftalmică progresivă a distrofiei musculare.
Până acum, nu au fost inventate mijloace care să ajute la scăderea completă a distrofiei în mușchi.
Există patru forme ale acestei patologii. Cel mai adesea, se pune diagnosticul de distrofie musculară Duchenne - în jumătate din toate cazurile de patologie. De obicei. Cursul bolii începe deja în copilărie și provoacă moartea până la vârsta de douăzeci de ani. Distrofia musculară Becker progresează ceva mai lent, pacienții care trăiesc până la vârsta de patruzeci de ani. Alte forme ale bolii de obicei nu au niciun efect asupra duratei vieții umane.
Factori etiologici care cauzează distrofie în mușchi
Formarea distrofiei în mușchi se datorează influenței diferitelor gene. Patologia Duchenne și Becker este cauzată de gene localizate pe cromozomii sexuali. Aceste forme sunt caracteristice doar pentru bărbați. Alte leziuni nu se corelează cu cromozomii sexuali, astfel încât atât bărbații, cât și femeile pot fi afectați de acestea.
Principalele manifestări și semne ale progresiei bolii
Toate tipurile de distrofie musculară provoacă dezvoltarea activă a atrofiei musculare, dar pot varia în funcție de severitatea patologiei și de momentul formării acesteia.
- Distrofia Duchenne se manifestă deja în copilăria timpurie - aproximativ între trei și cinci ani. În același timp, pacienții merg într-o epavă, este dificil să urceți scări, adesea servesc din senin și nu pot alerga. Când un copil cu un astfel de diagnostic ridică brațele, omoplații par să se îndepărteze de corp. Un copil cu acest tip de distrofie este închis într-un scaun cu rotile până la vârsta de 10-12 ani, iar slăbirea constantă progresivă a mușchilor provoacă moartea prin insuficiență cardiacă bruscă, insuficiență respiratorie sau leziuni infecțioase.
- Distrofia Becker are un număr mare de asemănări cu tipul anterior de patologie, dar progresează mult mai lent. Simptomele distrofiei musculare încep să apară abia la vârsta de cinci ani, iar după cincisprezece ani, pacienții pot încă să meargă independent, uneori chiar mult mai mult.
- Forma omoplatului-facială a distrofiei musculare progresează foarte lent, cursul său este relativ benign. Practic, boala se face simțită la vârsta de 10 ani, dar se poate manifesta și la începutul adolescenței. Copiii cu acest diagnostic deja în copilărie nu sug bine, iar la o vârstă mai înaintată nu își pot plia buzele într-un tub, își ridică mâinile deasupra capului. Fața este caracterizată de expresii faciale slabe în timpul plânsului sau al râsului, dar expresiile faciale sunt încă prezente uneori, în ciuda acestui fapt, este foarte diferită de normal.
În centrele de îngrijire medicală, care sunt dotate cu cea mai modernă tehnologie pentru realizarea examenelor imunologice și moleculare, specialiștii pot determina cu exactitate dacă copilul va suferi de distrofie musculară în viitor. În astfel de instituții, se organizează examinări și pentru părinții și rudele copilului și se dezvăluie prezența genelor în ele, care determină formarea distrofiei musculare Becker sau Duchenne.
Cum se desfășoară procesul de vindecare?
În medicina modernă, metodele de prevenire sau de progresare a dezvoltării active a acestei patologii nu au fost încă dezvoltate. Tratamentul distrofiei musculare presupune organizarea opoziției la complicații, de exemplu, deformări vertebrale datorate mușchilor slabi ai spatelui, tendința organismului de a contracta pneumonie din cauza slăbirii mușchilor respiratori.
Pacienților cu dezvoltare suplimentară a blocului cardiac li se poate implementa implantarea unui stimulator cardiac. Pentru tratamentul leziunilor cardiace este recomandat medicamentul fenigidin. Recepția diferitelor ortopedice vă permite să întăriți picioarele suspendate, să restabiliți funcționarea articulațiilor gleznelor și, de asemenea, să reduceți incidența căderilor.
Antrenamentul selectat corespunzător are, de asemenea, un efect pozitiv asupra cursului patologiei. Odată cu dezvoltarea atrofiei, grupurile anabolice de steroizi sunt utilizate pentru tratament, precum și pentru terapia de restaurare. Cu o manifestare puternică a simptomelor miotonice ale leziunii, este prescris un curs de tratament cu difenin care durează două până la trei săptămâni. Difenin este cel care probabil inhibă efectul patologic asupra conducerii sinaptice și, de asemenea, reduce activitatea musculară post-tetanică. Medicamentul Selegin dă rezultate pozitive atunci când este luat pentru a corecta tiparele de somn și pentru a elimina somnolența ridicată. 
Terapia eficientă poate fi efectuată numai datorită terapiei genice, care este în prezent dezvoltată activ. Un număr mare de lucrări experimentale indică o îmbunătățire a stării fibrelor musculare în tratamentul anumitor forme de boală. Odată cu dezvoltarea distrofiei Becker și Duchenne, există o producție insuficientă de proteină musculară - distrofină. Gena care este responsabilă de formarea acestei proteine este cea mai mare genă cunoscută în medicină, în legătură cu care, oamenii de știință au recreat o versiune mini a acestei gene, iar adenovirusurile au devenit cei mai buni conducători ai genei în mușchi.
Lista distrofiilor musculare progresive (PMD) include miodistrofia:
- pseudohipertrofic;
- Becker-Kener pseudohipertrofic;
- Emery-Dreyfus-Hogan;
- Rottaufa (miopatie fibrozată);
- juvenil Erba-Rota;
- oculară (oftalmoplegia Gref);
- umar-scapular-facial (Landuzi);
- oculofaringian;
- Dreyfus;
- mitocondriale.
Cauzele distrofiilor musculare
La pacienții cu PMD, este detectat un defect structural congenital în țesutul muscular (de exemplu, în Duchenne PMD, un defect al genei responsabile de sinteza proteinei musculare structurale distrofinei). Spre deosebire de pierderea acestei proteine în miodistrofia Duchenne, în miodistrofia Becker, distrofina este alterată calitativ. Pacienții cu PMD au:
- excitabilitatea și conductivitatea fibrelor musculare;
- microcirculația;
- influențe neurotrofice;
- metabolismul muscular.
Factorii provocatori sunt, în special, infecțiile, intoxicațiile, leziunile (fizice și/sau psihice) și bolile somatice.
Simptomele distrofiilor musculare
Simptome comune ale PMD:
- slăbiciune musculară (simetrică);
- absența durerii permanente;
- manifestare mai frecventă a slăbiciunii în secțiunile proximale, predominanța sa în mușchii pelvinului, centurii scapulare;
- scăderea şi stingerea reflexelor tendinoase proporţional cu slăbiciunea musculară.
În general, caracteristicile clinice ale formelor familiale și sporadice de miopatii sunt similare. Progresează încet și treptat. Localizarea atrofiei în diferite forme de miopatii:
- umăr;
- pelvin;
- pelvin-umăr;
- omoplat-facial;
- distal;
- ochi;
- oculo-bulbar;
- amestecat.
Distribuția distrofiei musculare este ascendentă sau descendentă. Mersul capătă așa-numitul caracter de rață. Din poziția culcat, pacienții se ridică cu ajutorul unor mișcări suplimentare - tehnici miopatice. Alături de atrofie, se observă și pseudohipertrofie a mușchilor (la 37% dintre pacienți), în principal la mușchii gastrocnemius și cvadriceps, mai rar la mușchii deltoid, supraspinatus, infraspinatus și intercostali. Slăbiciunea musculară crește odată cu dezvoltarea bolii și duce la o scădere a forței musculare, care în etapele ulterioare este estimată la 0-1 punct (conform unui sistem de cinci puncte). Concomitent cu creșterea severității atrofiei mușchilor scheletici, se observă o scădere și dispariție a reflexelor periostale ale tendonului. La majoritatea pacienților (83,5%) se determină modificări vegetativ-vasculare: hiperhidroză, acrocianoză a picioarelor și mâinilor, labilitate crescută a vasomotorilor, dermografie roșie persistentă. Pentru diferite forme de miodistrofie primară, sunt caracteristice și unele simptome și fenomene comune.
Miodistrofie pseudohipertrofică Duchenne
Distrofia musculară Duchenne pseudohipertrofică este o formă malignă de PMD. Boala este moștenită legată de cromozomul X. În miodistrofia Duchenne, a fost identificată o absență determinată genetic a proteinei musculare structurale distrofină, ceea ce duce la lansarea unei cascade de reacții chimice care provoacă moartea miofibrilelor. În conformitate cu acest tip de moștenire a bolii, băieții sunt de obicei bolnavi, ale căror mame sunt purtătoare ale genei recesive. Cu distrofia musculară Duchenne, există un efect de bunic: de la bunic, boala se poate transmite prin fiică la nepot.
Primele semne de distrofie musculară apar în primul an, iar până la sfârșitul acestuia, decalajul de dezvoltare a copiilor devine vizibil. De la vârsta de 2 ani se remarcă slăbiciune musculară.
Manifestări principale
Mers „de rață”.
În timpul mersului, pacientul „se rostogolește” de la picior la picior, ceea ce se datorează slăbiciunii mușchilor fesieri.
Pseudohipertrofie - „caviarul gnom”
Pseudohipertrofia vițeilor datorită infiltrației lor grase, proliferarea formațiunilor de țesut conjunctiv. Mușchii sunt bombați, fermi la atingere, dar puterea este redusă (un semn al miopatiei Duchenne)
"Pântec de broasca"
În unele forme de patologie neuromusculară, tonusul muscular scăzut duce cu distrofie musculară la faptul că abdomenul iese înainte.
Simptom de „brânză scapulară lentă”
Dacă copilul este crescut luându-și axilele, atunci antebrațele se ridică brusc, iar capul pacientului „se scufundă” între ele - un semn de hipotensiune severă a mușchilor centurii scapulare.
Zâmbet „transvers”.
Slăbiciunea și hipotrofia mușchilor faciali pot fi însoțite de o modificare a expresiilor faciale, în special de o întindere transversală a gurii. Semn al unor forme de miopatie.
Simptomul Shereshevsky-Govers
Un pacient cu miopatie, ridicându-se dintr-o poziție în decubit, face o serie de mișcări (se întoarce pe burtă, se pune în patru picioare), apoi începe să se ridice, dându-și treptat picioarele și sprijinindu-se pe mâini; mâinile își schimbă constant poziția, în timp ce pacientul „se urcă” pe propriile picioare, ca pe o scară.
Triada de sindroame în distrofia musculară: trei „A”
- Atrofie (hipotrofie)
- atonie (hipotensiune)
- Areflexie (hiporeflexie) a centurii pelvine, rezultând un mers „de rață”.
Răspândirea caracteristică în distrofia musculară a procesului miodistrofic este ascendentă - în timp, toți mușchii trunchiului și ai centurii scapulare sunt implicați în proces. Pieptul este turtit, se notează scolioza regiunii toracice și hiperlordoza lombară. Până la vârsta de 7 ani, pacienții au dificultăți în mișcare, până la vârsta de 12-15 ani își pierd capacitatea de a merge.
Fenomenele Shereshevsky-Govers, hiperlordoza lombară, pseudohipertrofia grupurilor musculare individuale sunt caracteristice. Pseudohipertrofia gambelor („viței pitici”) este tipică; la ridicare, pacienții întâmpină dificultăți severe. Odată cu implicarea mușchilor centurii scapulare, se formează omoplați „în formă de aripă”, se formează cifoscolioză, se dezvoltă slăbiciune a brațelor și a mușchilor respiratori.
În etapa târzie a procesului miodistrofic, apare hipotrofia mușchilor feței, faringelui și laringelui; exista contractura de flexie in articulatiile extremitatilor. Se dezvoltă cardiomiopatia (extinderea limitelor inimii, aritmii cardiace), sunt posibile modificări ale electrocardiogramei (ECG), sindrom adipozogenital, hipoplazie suprarenală și osteoporoză.
Aproximativ o treime dintre pacienți (30%) sunt în urmă în dezvoltarea funcțiilor intelectuale. În stadiul incipient, activitatea CPK, precum și activitatea LDH, crește brusc (de zeci și sute de ori).
Miodistrofie pseudohipertrofică Becker-Kener
Distrofia musculară pseudohipertrofică Becker-Kener este considerată o formă ușoară de Duchenne PMD. Boala se transmite, de asemenea, legat de cromozomul X. Debutul bolii se notează la vârsta de 5 ani.
Cursul procesului miodistrofic este lent progresiv. Caracteristicile distribuției distrofiilor musculare sunt identice cu semnele patologiei musculare la Duchenne PMD. Patologia mai puțin pronunțată a inimii. Inteligența pacienților este salvată; își păstrează capacitatea de a se mișca independent pentru o lungă perioadă de timp, pot avea copii.
Miodistrofie Emery
Distrofia musculară Emery-Hogan este moștenită legată de X. Retractiile tendoanelor calcaneale se dezvoltă precoce, la mers, se observă sprijinul pe degete, precum și manifestarea unui mers „de rață”. Se notează distrofia miocardică, contracturi multiple ale articulațiilor mari, rigiditatea coloanei vertebrale, piept în formă de butoi. Activitatea CPK este moderat crescută. Cursul este treptat progresiv. Inteligența salvată.
Miodistrofie Rottauf (fibrozatoare)
Debutul distrofiei musculare apare în copilărie, de obicei la vârsta de 4-12 ani. Există contracturi ale tendonului pronunțate. Se notează limitări ale flexiei dorsale a picioarelor, flexiei gâtului. Din cauza fibrozei musculare se formează posturi patologice, ducând la imposibilitatea flexiei coloanei vertebrale. Procesul miodistrofic progresează încet. Apar slăbiciune musculară și malnutriție moderată, mai mult în regiunea scapular-umăr. Inteligența salvată. dezvoltarea cardiomiopatiei. Hiperenzima severă este caracteristică. EMG dezvăluie modificări care indică un proces miodistrofic primar.
Miodistrofie juvenilă
În primul rând, boala se caracterizează prin atrofia mușchilor pelvieni. O manifestare precoce a bolii este mersul „de rață” (miopatic). Pacientul are dificultăți în a se ridica din poziție culcat. Se dezvăluie hiperlordoza lombară, abdomenul „broaștei”, atrofia membrelor superioare (forma Leiden-Moebius). Poate dezvoltarea unei pseudohipertrofii moderate. Pe fondul implicării în procesul mușchilor intercostali, diafragma, poate apărea insuficiență respiratorie.
Pacienții sunt susceptibili de a avea endocrinopatii (obezitate) și distonie autonomă. Cursul procesului miodistrofic este relativ ușor. În condiții nefavorabile (de exemplu, în timpul efortului fizic), este posibilă o progresie rapidă a procesului.
La EMG cu distrofie musculară, există o imagine a procesului miodistrofic primar. Pe ENMG, viteza pulsului se încadrează în norma de vârstă. În sânge se determină hiperfermentemie moderată.
Miodistrofia umar-scapular-facial (Landuzi)
Boala este moștenită în mod autosomal dominant. Se presupune că gena patologică este localizată pe cromozomul 4. Există o penetranță pronunțată a genei.
Debutul bolii are loc de obicei la vârsta de 20 de ani, boala începe cu slăbiciune a mușchilor feței. Observat timpuriu „buzele unui tapir”, zâmbetul Giocondei). În timp, scăderea în greutate și slăbiciunea mușchilor serratus anterior și pectoral major cresc. Mai târziu, procesul miodistrofic afectează grupul muscular peronier, apare un mers „steppage”. Se dezvoltă pseudohipertrofie moderată a mușchilor. Metabolismul creatin-creatinină este moderat perturbat.
Miodistrofie oculară (Grefe)
Boala debutează înainte de vârsta de 30 de ani. Deteriorarea mușchilor oculari crește, care se desfășoară fără diplopie, duce la imobilitatea globilor oculari (paralizia privirii). La început, mușchiul care ridică pleoapa superioară este adesea afectat; în stadiul târziu al procesului miodistrofic se observă ptoză bilaterală. Uneori sunt implicați mușchii mimici, bulbari și scheletici.
Miodistrofie oculofaringiană
Miodistrofia oculofaringiană este o oftalmoplegie externă clasică cu disfagie și disfonie, moștenită în mod autosomal dominant. În 1961, boala a fost descrisă la reprezentanții populației franco-canadiene.
Varianta clasica a bolii este oftalmoplegia progresiva cu disfagie si disfonie, insotita de ptoza pleoapelor superioare. O altă variantă a bolii procedează cu adăugarea de pareză a mușchilor care asigură mișcările ochilor, mușchii faciali și de mestecat și mușchii gâtului. În a treia variantă, rară, în proces sunt implicați și mușchii membrelor. Activitatea enzimelor (CPK și LDH) este ușor crescută.
Miodistrofia Dreyfus
Distrofia musculara Dreyfus se manifesta prin cresterea slabiciunii musculare, in principal a centurii pelvine si a extremitatilor inferioare. Mersul caracteristic pacientului pe baza degetelor mari; hiperlordoză lombară marcată.
O caracteristică a acestei forme de miodistrofie este formarea de contracturi pronunțate ale cotului și ale altor articulații. Miocardul este adesea afectat, pacientul rămâne în urmă în dezvoltarea psihică.
Miopatii mitocondriale
În miopatiile mitocondriale, defectul biochimic este localizat în mitocondriile celulelor, care este detectat prin studii biochimice și ultramicroscopice. Cel mai adesea, debutul bolii are loc în a 2-a decadă a vieții.
În stadiile incipiente apar ptoza, oftalmopareza externă (fără diplopie din cauza simetriei mușchilor oculomotori), slăbiciune a mușchilor proximali, hipo- și areflexia tendonului.
Durata progresiei distrofiei musculare este variabilă: de la luni la zeci de ani. Studiile clinice și neurofiziologice (EMG, ENMG) în miopatiile mitocondrale relevă manifestări miodistrofice și neuropatice.
Diagnosticare
Diagnosticul necesită studii neurofiziologice, biochimice și histopatologice.
EMG local. La pacienții cu miopatii, interferența patologică este înregistrată cu o frecvență ridicată a potențialelor polifazice și o scurtare a timpului de formare a oscilațiilor individuale.
Cercetare biochimică. În stadiile incipiente, activitatea CPK în sânge crește de 50 de ori sau mai mult; LDH - de 5-7 ori; FDA - de 2-5 ori; în etapele ulterioare scade la norma de vârstă.
Studiu patologic. Semnele PMD sunt determinate:
- regenerarea țesutului muscular;
- aranjarea dezordonată a fibrelor musculare de diferite dimensiuni;
- alternarea fibrelor normale, atrofiate și (la unii mușchi) hipertrofiate;
- atrofia fibrelor musculare în lungime și diametru;
- proliferarea nucleelor cu localizarea lor sub sarcolemă și în interiorul fibrei.
Tratamentul distrofiilor musculare
Tratamentul primar are ca scop încetinirea ratei de dezvoltare a bolii și maximizarea capacității pacientului de autoservire.
Tactici pentru tratamentul PMD:
- terapie dietetică pentru a evita dezvoltarea excesului de greutate la un pacient;
- și activitatea motrică, în special, pentru menținerea funcțiilor articulațiilor de susținere și prevenirea contracturilor;
- exerciții de respirație;
- terapie medicamentoasă;
- reabilitare socio-psihologică;
- îngrijire ortopedică.
În unele forme de miodistrofie, în special în miodistrofia Duchenne, utilizarea glucocorticoizilor face uneori posibilă amânarea declanșării imobilității pacientului cu ani de zile. Deoarece un astfel de tratament este lung și de obicei însoțit de multe complicații, este necesară o administrare deliberată de glucocorticoizi. În PMD, se utilizează prednisolon 1-2 mg/kg dimineața, o dată la două zile (ținând cont de eventualele contraindicații), prednisolonul zilnic este posibil și pentru distrofia musculară (o dată dimineața la o doză de 0,75 mg/kg/zi). ). Autorii sugerează, de asemenea, utilizarea unui medicament din același grup - deflazacort, care are mai puține efecte secundare, la aproximativ aceeași doză (din punct de vedere al eficacității, 6 mg deflazacort corespund la 5 mg prednisolon). Durata tratamentului depinde de eficacitatea și severitatea efectelor secundare ale medicamentului.
Este prezentată terapia simptomatică și restaurativă. În special, se recomandă utilizarea de adenozinfosfat, trifosadenină, vitamina E, coenzime (de exemplu, cocarboxilază), agenți anabolizanți non-hormonali (etiltiobenzimidazol, acid orotic), inozină, preparate cu potasiu. În același timp, trebuie avută în vedere lipsa dovezilor manifestărilor obiective ale eficacității acestor medicamente.
Prednisolon în interior (dimineața) 1-2 mg/kg/zi la două zile, durata terapiei se stabilește individual, sau Prednisolon în interior (dimineața) 0,75 mg/kg/zi zilnic, pe termen lung sau Deflazacort în interior 6 mg, pe termen lung.
Prognosticul distrofiilor musculare
Prognosticul miopatiilor ereditare depinde de formă. Deoarece la majoritatea pacienților boala este progresiv progresivă, prognosticul este prost.
Pacienții cu miodistrofie Duchenne pseudohipertrofică mor de obicei în decada a 3-a de viață, mai des din cauza insuficienței cardiace progresive. Cu distrofia musculară pseudohipertrofică a lui Becker-Kener și distrofia musculară a lui Emery-Dreyfus-Hogan, pacienții trăiesc de obicei până la 40-60 de ani. Cu miodistrofia Rottauf-Mortje-Beyer, pacienții trăiesc adesea până la 30-50 de ani.
Articolul a fost pregătit și editat de: chirurgDistrofia musculară este un grup de boli care provoacă degenerarea mușchilor scheletici și a articulațiilor. Medicamentele pentru tratamentul acesteia nu există încă, deși există proceduri medicale eficiente care pot încetini dezvoltarea bolii. Simptomele pot fi ușoare, dar în cazurile severe, boala afectează reflexele și face pacientul să nu poată merge.
Varietăți de distrofie musculară
Au fost identificate peste 30 de tipuri de distrofie musculară, dintre care 9 sunt cele mai frecvente:
- Distrofia musculară Duchenne - cea mai frecventă formă, se dezvoltă în principal la bărbați tineri;
- Distrofia lui Becker - până la vârsta de patruzeci de ani, pacientul își pierde capacitatea de a se mișca independent;
- miotonie - simptomele apar mai des la adulți (sau la copii în timpul copilăriei târzii);
- distrofie congenitala - diagnosticata imediat dupa nastere sau in primele luni de viata;
- limbus-belt - caracteristică adolescenților, tinerilor (20-25 de ani), afectează mușchii șoldurilor și umerilor;
- umăr-scapular-facial - provoacă slăbirea mușchilor feței, a mușchilor umărului, pierderea capacității de a ridica brațele, probleme cu vorbirea, tahicardie;
- distal - boala se caracterizează prin slăbiciune a brațelor și a extremităților inferioare;
- Distrofia Emery-Dreyfus - pentru femei este periculoasă cu paralizia cardiacă, la copii (în principal băieți), pe lângă inimă, sunt afectați mușchii gambei și centura scapulară superioară;
- Boala Charcot (și alte boli degenerative) poate provoca, de asemenea, simptome similare cu distrofia musculară.
Dintre aceste 9 forme de distrofie musculară, următoarele sunt cel mai frecvent diagnosticate:
- Distrofia Duchenne. Simptomele apar în copilăria timpurie (înainte de vârsta de 5 ani). Boala este diagnosticată la unul din 3.500 de băieți nou-născuți. Este cea mai frecventă tulburare genetică fatală. Femeile pot fi purtătoare ale unei gene care a suferit o mutație, dar ele însele nu suferă de această tulburare. Modificările degenerative blochează o proteină (distrofină) care susține elementele structurale ale țesutului muscular și ale membranelor celulare. Fără distrofină, slăbiciunea musculară progresează, ducând la imobilitate și moarte.
- Distrofia lui Becker. Deși producția de distrofină este păstrată în această boală, nivelul proteinei în formă de tijă este semnificativ sub normal. Distrofia musculară se dezvoltă până la vârsta de 12 ani. Boala se dezvoltă mai lent, dar leziunile sunt semnificative: curbura coloanei vertebrale, dificultăți de respirație, oboseală constantă, slăbiciune, boli de inimă, probleme cognitive.
Principalele simptome ale distrofiei
Deși toate tulburările sunt legate, fiecare tip de distrofie este cauzată de o mutație genetică unică, astfel încât simptomele și momentul bolii variază:
- pierderea masei musculare (problema se agraveaza cu varsta);
- înfrângerea extremităților inferioare (o slăbiciune suplimentară se extinde la mușchii gâtului, umerilor, spatelui, pieptului);
- epuizare progresivă;
- o creștere a mușchilor gambei și deltoizi;
- scăderea flexibilității, rezistenței, oboselii;
- încălcarea coordonării, sincronizarea mișcărilor;
- căderi (din cauza pierderii coordonării și a slăbiciunii musculare);
- rigiditate articulară, durere la mișcare;
- înlocuirea țesutului fibros cu mușchi;
- este dificil să te ghemuiești, să te apleci, să urci pe scări;
- dezvoltarea anormală a scheletului (curbura coloanei vertebrale, postură afectată);
- intervalul de mișcare scade (până la paralizie completă);
- se dezvoltă pneumonie, alte probleme cu funcționarea sistemului respirator, activitatea cardiacă este perturbată, ceea ce poate duce la moarte (cu distrofie Duchenne).
Distrofia este moștenită. Cu toate acestea, în 35% din cazuri, boala provoacă o mutație spontană a genelor din cauza unei încălcări a producției de proteine. Bărbații sunt expuși unui risc mai mare datorită faptului că au un cromozom X. Cromozomul Y (un alt cromozom sexual întâlnit la bărbați) nu are o copie a genei distrofinei și nu are niciun efect asupra dezvoltării distrofiei musculare.

La femei, riscul de distrofie este redus datorită prezenței a doi cromozomi X. Chiar dacă unul dintre cromozomi este anormal, o genă sănătoasă poate produce suficientă distrofină. Probabilitatea de a moșteni o genă defectuoasă este de 50%.
Diagnosticul distrofiei musculare
La diagnosticarea distrofiei, ei efectuează:
- examen fizic - se verifică flexibilitatea, se testează forța musculară, gama de mișcare și alte caracteristici ale pacientului;
- electromiografie - examinează activitatea electrică din interiorul mușchilor;
- electrocardiogramă;
- biopsie musculară - vă permite să determinați ce tip de distrofie se dezvoltă;
- imagistica prin rezonanță magnetică – arată care organe și țesuturi sunt afectate.
Femeile pot face un test atunci când plănuiesc o sarcină pentru a ști dacă gena defectă este prezentă sau nu. Un test de sânge poate determina prezența enzimelor asociate cu dezvoltarea anormală a mușchilor.
Tratamentul distrofiei musculare
Tratamentul depinde de tipul de distrofie. Distrofia Becker și Duchenne sunt incurabile, dar există modalități de a gestiona simptomele bolii. Cu cât sunt combinate mai multe metode (inclusiv dietă, terapie emoțională și fizică, suport cu medicamente), cu atât rezultatul este mai bun.
Terapia poate include:
- luarea de steroizi (pentru combaterea slăbiciunii musculare și ameliorarea simptomelor dureroase), albuterol (dacă este prezent astmul), medicamente care îmbunătățesc funcția cardiacă (angiotensină, beta-blocante), inhibitori ai pompei de protoni, suplimente alimentare pentru menținerea energiei și a masei musculare;
- utilizarea de produse ortopedice (brete, scaune cu rotile);
- tratamentul patologiei vorbirii (dacă sunt afectate fața și limba).

Metode naturale pentru gestionarea distrofiei musculare
Activitate fizica
Este important să încercați să mențineți flexibilitatea și forța musculară. Modul pasiv, absența sarcinilor elementare exacerbează simptomele bolii, contribuie la dezvoltarea complicațiilor și a suferinței emoționale. Kinetoterapie este utila pentru mentinerea flexibilitatii si coordonarii organismului. Mobilitatea poate fi mărită folosind incrustații ortopedice, un corset, un baston, un scuter electric, un scaun cu rotile și vă puteți deplasa cu sprijinul unui asistent.
De la durerile articulare, la îmbunătățirea echilibrului, reducerea anxietății, menținerea amplitudinii de mișcare, sunt utile înotul, yoga, exercițiile de întindere (s-au dezvoltat practici speciale pentru cei care suferă de distrofie musculară, le puteți găsi pe internet sau la medic).
Suport psihologic
Dieta sanatoasa
Unele substanțe chimice alimentare afectează genomul uman și modifică structura genelor, provocând apariția sau progresia bolilor cronice, dezvoltarea complicațiilor. Procesele inflamatorii sunt susținute de o dietă sărăcită în substanțe utile (sau supraîncărcate cu alimente procesate), lipsa de activitate fizică și stres. Acești factori reduc capacitatea organismului de a proteja celulele de degenerare, accelerează îmbătrânirea și duc la pierderea densității osoase.
O dietă antiinflamatoare, deși nu se va putea vindeca de distrofia musculară, va încetini semnificativ progresia bolii. O astfel de dietă reduce inflamația, alcalinizează organismul, scade nivelul de glucoză, ajută la eliminarea toxinelor și oferă nutrienți.
Principii de nutriție:
- înlocuirea grăsimilor „rele” cu cele „bune” - evitarea uleiurilor hidrogenate (soia, rapiță), a grăsimilor trans, introducerea grăsimilor saturate și nesaturate sănătoase (măsline, floarea soarelui, cocos, semințe de in, ulei de susan, avocado, grăsimi omega-3);
- alegerea produselor din carne organică (fără antibiotice, hormoni);
- excluderea zahărului rafinat și a glutenului.
Cele mai bune alimente antiinflamatoare sunt legumele cu frunze, bok choy, țelina, sfecla, broccoli, afinele, ananasul, somonul (sălbatic), bulionul de oase, nucile, turmericul, ghimbirul, semințele de chia.

Pe lângă apa pură, sunt utile ceaiurile din plante, sucuri proaspăt stoarse, limonada naturală, băutura din fructe, kvas. Din produsele lactate, este permis să lăsați în meniu lapte de capră (brânză, iaurt din el), brânză de oaie.
Este important să excludem posibilitatea ca toxinele să intre în organism nu numai din alimente, ci și din substanțele chimice de uz casnic, cosmetice, alegând remedii naturale.
Suplimente de sprijin muscular
Conform prescripției medicului, pentru distrofia musculară pot fi utilizate următoarele suplimente:
- aminoacizi (carnitina, coenzima Q10, creatinina) - contribuie la producerea proteinelor necesare sustinerii musculare;
- glucozamină și condroitina - combate durerile articulare;
- antioxidanti (vitaminele C, E, A) - buni pentru inima, articulatii si muschi;
- ceai verde, matcha (extracte) - susțin nivelul de energie;
- acizi omega-3, ulei de pește - reziste la inflamație;
- prebiotice - îmbunătățesc digestia.
Uleiuri esentiale
Uleiul de mentă, tămâie, ghimbir, turmeric, smirnă ajută la reducerea umflăturilor, ameliorarea durerii și a altor simptome cauzate de degenerarea mușchilor, țesuturilor și articulațiilor. Eficient în tratamentul depresiei, pentru ameliorarea anxietății ulei de lavandă, mușețel, grepfrut, lemn de santal. Puteti adauga ulei picatura cu picatura in difuzorul unui umidificator, faceti bai pe baza acestuia si folositi-l pentru masaj.
Dacă se găsesc semne alarmante (amorțeală facială, tulburări de vorbire, mers, slăbiciune bruscă, flexibilitate redusă), este necesar să se supună unei examinări. Intervenția precoce va încetini progresul bolii și va reduce impactul negativ al simptomelor distrofiei musculare asupra calității vieții.
În neurologia modernă, există un număr mare de boli, a căror natură apariției nu poate fi explicată rațional de către specialiști. Astfel de boli includ un grup de afecțiuni precum distrofia musculară. Există nouă varietăți ale acestei boli, dar mai întâi de toate...
Distrofia musculară este o boală ereditară cronică care duce la deteriorarea sistemului muscular uman. Mușchii afectați încetează să funcționeze normal, devin mai subțiri în dimensiune și un strat de grăsime crește treptat la locul lor în corp.
Varietăți de distrofie musculară
În neurologia modernă, această boală este clasificată în nouă boli diferite. Clasificarea bolii este asociată cu:
- localizarea tulburărilor musculare;
- caracteristicile bolii;
- dezvoltare agresivă;
- vârstă.
Deci, se întâmplă distrofia musculară:
- Duchenne;
- miotonic (boala Steinert);
- Becker;
- Erba Roth;
- forma juvenilă de distrofie Erba-Roth;
- umăr - formă facială scapulară (Landuzi-Dejerine);
- miopatie alcoolică;
- forma distală;
- Miodistrofia Emery-Dreyfus.
Distrofia Duchenne
Cea mai cunoscută formă de distrofie Duchenne progresivă (distrofie pseudohipertrofică, merozină - distrofie congenitală negativă). Acest tip de boală se manifestă în copilărie de la doi la cinci ani. În primul rând, mușchii extremităților inferioare suferă de această boală, care, în ciuda unui stil de viață sedentar la pacienții tineri, crește treptat în dimensiune. Această caracteristică este asociată cu o creștere a țesutului adipos în loc de mușchi.

Copil sănătos în stânga, copil bolnav în dreapta
Treptat, pe măsură ce boala progresează, se deplasează în partea superioară și afectează mușchii membrelor superioare. De regulă, până la vârsta de doisprezece ani, un pacient mic încetează complet să se miște. Letalitatea acestei boli este foarte mare, aproximativ 85-90% dintre pacienți nu trăiesc până la 20 de ani.
Bărbații sunt expuși riscului, deoarece această boală practic nu afectează fetele.
boala Steinert
Nu degeaba distrofia congenitală a lui Steiner se numește miotonică, deoarece, ca urmare a progresiei bolii, mușchii se relaxează prea încet după contracția lor (acest fenomen se numește miotonie).
Această boală, spre deosebire de cea anterioară, este frecventă la adulții cu vârsta cuprinsă între 20 și 40 de ani. Există și cazuri de progresie a bolii la copii, de obicei în copilărie, cu toate acestea, aceasta este mai degrabă excepția decât regula.

Semne faciale de miotonie (gura și pleoapele deschise)
Boala nu are o dependență de gen și afectează în egală măsură atât bărbații, cât și femeile. Se observă că, în cazul unei boli, se manifestă slăbiciune a mușchilor faciali ai feței, precum și a membrelor. Progresia, spre deosebire de boala Duchenne, este lentă.
O trăsătură distinctivă a bolii este posibilitatea de deteriorare nu numai a mușchilor membrelor, ci și a mușchilor interni (mușchiul inimii), care, la rândul său, prezintă un mare pericol pentru viața umană.
boala Becker
Această formă a bolii progresează de asemenea pentru o lungă perioadă de timp, iar pacientul se simte bine pentru o lungă perioadă de timp. O exacerbare a bolii poate apărea pe fondul leziunilor sau diferitelor boli ale sistemului nervos, care, odată cu cursul lor, vor accelera dezvoltarea bolii.
La risc sunt persoanele de statură mică.
Boala Erb-Roth și forma sa tinerească
Această boală autozomal recesivă se dezvoltă la pacienții după vârsta de 20 de ani, iar forma tinerească la copii și adolescenți cu vârsta cuprinsă între 11-13 ani. Progresia bolii are loc într-o variantă ascendentă, adică mai întâi sunt afectați mușchii extremităților inferioare, iar boala urcă treptat către extremitățile superioare. 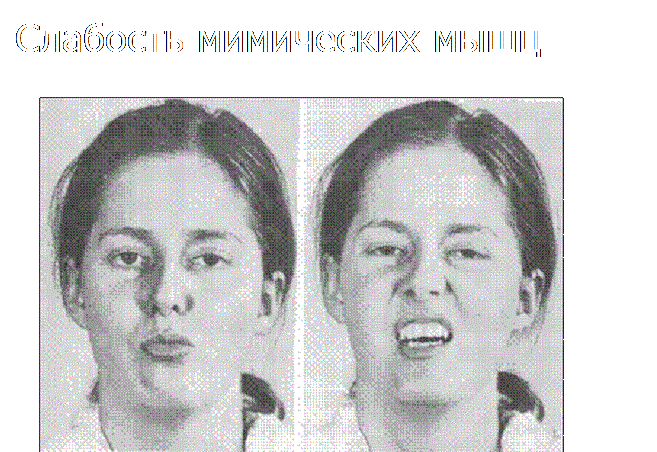
O trăsătură distinctivă a bolii este prezența omoplaților proeminenti, care, pe măsură ce progresează, devin mai pronunțate și mai evidente.
În timpul mersului, se notează transbordarea pacientului, proeminența abdomenului și retragerea toracelui.
boala Landouzy-Dejerine
Forma omoplat-facială a acestei boli este cea mai nediscriminatorie, deoarece afectează persoanele cu vârsta cuprinsă între cinci și 55 de ani. Această boală se caracterizează printr-o dezvoltare foarte lungă, pacientul putând rămâne capabil să lucreze până la 25 de ani de viață cu această boală.
Simptomele distinctive sunt afectarea mușchilor faciali, în urma cărora pacientul poate avea probleme cu claritatea pronunției, din cauza închiderii incomplete a buzelor. În plus, se observă închiderea incompletă a pleoapelor.
Pe măsură ce pacientul se dezvoltă, mușchii feței, mușchii umerilor, membrelor și trunchiului se atrofiază.
Această formă nu depinde de o mutație genetică.
Miopatie alcoolică
De asemenea, acest tip de boală nu este asociat cu mutații genetice și există un singur motiv pentru apariția sa - abuzul de alcool. Pacienții pot prezenta sindroame dureroase la nivelul extremităților asociate în mod specific cu leziuni musculare.
Există miopatie alcoolică acută și cronică.
Forma distală
Forma distală a distrofiei musculare este o variantă benignă a distrofiei progresive. De regulă, această boală este dificil de diferențiat de amiatrofia neuronală a lui Marie-Charcot. Pentru a separa aceste două boli, este necesară o electroencefalogramă a capului, care oferă o înțelegere a bolii care trebuie tratată.
Principalele simptome ale bolii includ atrofia mușchilor membrelor cu emaciarea lor ulterioară. Posibilă pareză a picioarelor, mâinilor etc.
Miodistrofia Emery-Dreyfus
Acest tip de boală nu a fost inițial identificat ca o boală separată, deoarece în simptomele sale era similar cu distrofia Duchenne. Cu toate acestea, mai târziu, în urma unui studiu pe termen lung, s-a constatat că boala Emery-Dreyfus are simptome individuale.
Boala este clasificată ca fiind rară. La risc sunt persoanele sub 30 de ani, de obicei o vârstă fragedă. Există dovezi ale manifestării simptomelor după 30 de ani, dar acestea sunt rare.
Principala diferență între acest tip de boală este problemele cu mușchii inimii, care în cele din urmă pot provoca moartea. Cardiomiopatia în această boală nu este singura diferență. Pe lângă problemele cardiace, pacienții au semne standard pentru distrofia Duchenne, dar într-un mod de dezvoltare mai benign.
Motivele
Componenta negativă a bolilor sistemului nervos este că sunt greu de studiat. Din acest motiv, nu se cunosc pe deplin factorii care provoacă dezvoltarea unei forme sau alteia de distrofie musculară.
Motivul principal pentru formarea majorității subspeciilor acestei boli este o mutație genetică, în special gena care este responsabilă de sinteza proteinelor.
De exemplu, boala Duchenne este direct legată de mutația cromozomului X sexual. Principalul purtător al unei gene rele sunt fetele care, în ciuda prezenței acesteia în propriul ADN, nu suferă de această boală.
În ceea ce privește forma miotonică, vinovată de apariția acesteia este o genă situată pe cromozomul 19.
Principalele simptome
Prezența unui număr mare de subspecii diferite în această boală indică diferențe de simptome, cu toate acestea, boala are simptome comune, care includ:

Diagnosticul distrofiei musculare
Măsurile de diagnosticare pentru o astfel de boală sunt extinse, deoarece există un număr mare de boli de care este necesar să se diferențieze boala.
În etapa inițială, medicul va studia cu siguranță istoricul pacientului, va clarifica simptomele, rutina zilnică etc. Aceste date sunt necesare pentru a elabora un plan pentru măsurile de diagnostic ulterioare, care pot include:

Este de remarcat faptul că, cu cât boala se manifestă mai târziu, cu atât este mai bine pentru pacient, deoarece simptomele precoce se termină în majoritatea cazurilor cu moartea.
Tratament
Tratamentul distrofiei musculare este un proces complex și de durată, totuși, în prezent, nu a fost creat un medicament care să vindece complet pacientul. Toate activitățile sunt menite să ușureze viața pacientului și să restabilească unele dintre abilitățile pierdute.
Pentru a încetini dezvoltarea bolii, sunt prescrise următoarele medicamente:
- corticosteroizi;
- vitaminele B1;
- adenozin trifosfat (ATP).
În plus, pentru a încetini procesul de dezvoltare, se folosesc celule stem fetale, care încetinesc procesul de distrofie.
În plus, următoarele sunt prescrise ca măsuri preventive:
- masaj;
- fizioterapie;
- exerciții de respirație.
Pe lângă opțiunile de tratament standard, este important să fii ghidat în mod constant de trei componente principale în procesul vieții:
- Activitate fizică adecvată.
- Suport psihologic în timp util.
- Tine dieta.
Activitate fizica
Lipsa dorinței unei persoane de a lupta împotriva bolii are un efect negativ asupra organismului. Judecă singur, pasivitatea, lipsa de dorință de a se mișca deprimă sistemul muscular deja afectat. Muschii trebuie să primească o sarcină, deoarece fără încărcare, procesele distrofice încep să apară mai repede, progresând astfel într-un ritm mai rapid.
Activitate fizică moderată, utilizarea dispozitivelor de susținere va fi un ajutor excelent în lupta împotriva bolii.
În prezența durerii în mușchi, înotul, yoga, exercițiile de întindere sunt perfecte.
Suport psihologic
Suportul psihologic al mediului este important pentru o persoană bolnavă. Și dacă boala este la fel de gravă ca aceasta, cu atât mai mult. Pentru unii, sprijinul obișnuit de la prieteni și familie va fi suficient, în timp ce alții ar putea avea nevoie de ajutor psihologic calificat.
Este important să-i spunem clar unei astfel de persoane că nu a rămas singur cu problema lui. Trebuie să înțeleagă că are la cine să apeleze, sunt oameni care îl empatizează și îl susțin.
Dietă
În ceea ce privește alimentația și dietă, există o credință comună că respectarea unei diete antiinflamatorii poate încetini progresia bolii. Această dietă reduce inflamația, nivelul de glucoză, elimină toxinele din organism și îl hrănește cu substanțe benefice.
Esența unei astfel de diete este următoarea:
- Refuzul produselor care conțin grăsimi „rele” și înlocuirea lor cu altele „bune”, introducerea în alimentație a grăsimilor nesaturate, care se găsesc în măsline, semințe de in, ulei de susan, avocado.
- Utilizarea cărnii și a peștelui în alimente, în producția cărora nu s-au folosit antibiotice sau hormoni.
- Eliminarea completă a zahărului rafinat și a glutenului din dietă.
- Mananca urmatoarele alimente - varza chinezeasca, broccoli, telina, ananas, somon, sfecla, cucumaria, ghimbir, turmeric si alte alimente care au proprietati antiinflamatorii.
- Produsele lactate sunt permise numai pe bază de lapte de oaie și capră.
- Este permisă utilizarea ceaiurilor din plante, limonadă, kvas, băuturi din fructe și sucuri naturale.
Prevenirea
Deoarece distrofia musculară este destul de dificil de prezis și detectat într-un stadiu incipient, măsurile preventive se reduc la două recomandări simple:
Examinarea obligatorie a unei femei în etapa de planificare a sarcinii pentru prezența mutațiilor în gene
Dacă, dintr-un motiv oarecare, nu a fost efectuat un examen genetic înainte de sarcină, un test este efectuat deja în timpul sarcinii pentru a determina mutațiile cromozomului X la făt.
Prognostic și complicații
În funcție de tipul bolii, prognosticul poate fi diferit și, cu toate acestea, se pot distinge mai multe posibile complicații care apar cu această boală.
- tulburări cardiace
- rahiocampsis
- scăderea abilităților intelectuale ale pacientului
- scăderea activității motorii
- tulburări ale sistemului respirator
- rezultat letal
Deci, distrofia musculară este o boală destul de periculoasă și incurabilă, așa că viitorii părinți trebuie să fie profund responsabili pentru planificarea sarcinii. Ai grijă de tine și de viitorii tăi copii!
Adevărul este că M-am uitat la microscopul electronic și am văzut(prezentat în alb în imagine) între fibrele musculare(Culoare rosie).
Pe imagine: biopsie de fibre musculare pentru miopatie ușoară (A), moderată (B) și severă (C):
Pe imagine: fibre musculare normale ale unei persoane sănătoase:

Pe exemplul pacientului meu care a suferit. Diagnosticul lui Emine: distrofie musculară severă biopsie confirmată. În continuare, voi descrie abordarea mea pentru eliminarea slăbiciunii musculare. Recomand să vizionați un videoclip despre tratamentul distrofiei musculare Duchenne progresive.
Distrofia musculară este o boală a unei încălcări a creării unei proteine care creează cadrul unei celule musculare.
- În cadrul celulei se formează găuri. Aceste găuri scurg compuși vitali și oligoelemente. Pentru a plasa găurile, celula este forțată să producă substanțe care sunt mai mari decât aceste găuri. Celula „se umflă” din interior, adică. se umfla.
- Creșterea edemului pune presiune asupra celulelor musculare din exterior, împingând nucleii celulari și mitocondriile spre periferie.
- În celulă, nivelul creatin fosfokinazei crește, iar mușchiul își pierde capacitatea de a lega și reține creatina.
- Creatina este necesară de mitocondrii pentru producerea de energie în celula musculară.
- Mitocondriile scad producția de ATP. ATP este energia necesară pentru a rula proteinele motorii actina și miozina. Fără energie - fără mișcare.
- În interiorul fibrei musculare, care nu se mișcă, propriile procese nutriționale încetinesc.
- Membrana fibroasă începe să secrete enzime și aminoacizi care nu sunt necesari pentru ea fără funcția de mișcare. Prin urmare, a apărut teoria „membranelor defecte”.
- În timpul mișcării musculare, sunt necesare aceste enzime și aminoacizi. Sinteza lor necesită energie, care nu este disponibilă. Prin urmare, apare slăbiciune musculară.
- Începe atrofia fibrelor musculare.
Simptome
Boala distrofia musculară începe cu dezvoltarea slăbiciunii și atrofiei unui anumit grup muscular. De-a lungul anilor, procesul distrofic captează din ce în ce mai multe grupuri musculare noi. Acest lucru se întâmplă până la imobilitate completă. Principalul simptom al miodistrofiei este înfrângerea mușchilor pelvini, ai centurii umărului și a trunchiului pacientului. Mușchii coapsei și mușchii umărului sunt afectați în cazurile severe, ceea ce a fost cazul pacientului Emine: nu se putea ridica fără sprijin și merge, nici măcar pe distanțe scurte.
Distrofii musculare bilaterale
În perioada inițială, miodistrofia, pe de o parte, poate predomina, dar pe măsură ce boala se dezvoltă, gradul de deteriorare devine același în mușchii simetrici ai pacientului. În timp, cursul bolii în aproape toți mușchii le scade puterea musculară. Pe corpul unui pacient care suferă de distrofie musculară apar zone de mușchi hipertrofici. Aceasta este pseudohipertrofie, care nu este asociată cu o creștere a fibrelor musculare. Pseudohipertrofia musculară este asociată cu umflarea mușchilor picioarelor sau brațelor. Astfel de mușchi sunt denși, dar slabi.
Forme de distrofie musculară la adulți
Toate formele figurative ale bolii în cauză la adulți diferă:
- tipuri de moștenire;
- viteza și natura cursului său;
- prezența sau absența retractiilor de tendon și a pseudohipertrofiilor;
- momentul începerii procesului;
- originalitatea topografiei suferinței musculare;
- alte semne ale distrofiei musculare progresive.
Întrebările de clasificare a miopatiei (boli musculare ereditare cronice și progresive) sunt dezvoltate în direcții diferite. Distrofia musculară la adulți este clasificată în funcție de tipul de moștenire:
- Autozomal dominant.
- Autosomal recesiv.
- Dominant și recesiv.
- Legat de cromozomul X.
Examinare pentru miopatie
Semnele clinice caracteristice distrofiei musculare sunt simptome de paralizie flască în diferite grupe musculare ale unei persoane bolnave, fără semne de afectare a neuronilor motori și a nervilor periferici. neurologi lumea întreagă nu poate explica asta.
Doctor Nikonov
Opinia mea: umflarea proteinelor între fibrele musculare face imposibilă mișcarea mușchilor.
Necunoașterea acestui fenomen îi perplexă pe medicii din întreaga lume: „Cum așa? Fibra musculară este intactă, nu este deteriorată. Neuronii motori și nervii periferici sunt întregi, la locul lor și trec perfect impulsurile care vin de la creier la mușchi și de la mușchi la creier, dar mișcările sunt dificile?
neurologi ordonat să facă electromiografie. Și din nou, un mister pentru ei: nu există modificări în structura fibrei musculare. O scădere a amplitudinii răspunsului M, interferență crescută și potențial polifazic indică dificultate în mișcarea musculară fără nicio patologie!
Tabloul anatomic patologic al bolii
Să aruncăm o privire la ceea ce se întâmplă în interiorul celulelor musculare la pacienții cu distrofie musculară Duchenne. Pentru a face acest lucru, vom face o incizie în piele, o vom extinde cu un expandator și vom lua o mică bucată de fibre musculare.


Un semn tipic de miodistrofie este, în primul rând, un diametru diferit al fibrelor musculare. La o persoană sănătoasă, diametrul fibrelor musculare este același.
Semnele caracteristice ale distrofiei musculare sunt fibrele atrofiate și hipertrofiate, nucleii interni multipli și edemul.
Examinând secțiuni colorate ale mușchiului scheletic, am văzut denervarea miofibrilelor, variații semnificative în dimensiunea miofibrilei și edem marcat.

Explicație pentru prima fotografie:
- Culoare violet pal - acestea sunt fibre musculare în context.
- Petele de lumină atât în interiorul fibrelor, cât și în exterior se umflă.
- Punctele întunecate sunt nucleele pe care edemul i-a deplasat la periferie.
Pe a doua fotografie este prezentată fibra musculară normală a unei persoane sănătoase.
Severitatea distrofiei musculare Conform microscopiei electronice, se concentrează pe următorii indicatori:
- la grad ușor diferența de dimensiune a fibrelor musculare este moderată, semnele inițiale de edem (culoare albă).
Pe imagine: biopsie de fibre musculare pentru distrofie ușoară (A), moderată (B) și severă (C).
- grad mediu severitatea corespunde mișcării nucleelor spre centrul fibrelor musculare, extinderii spațiului interfibrilar datorită creșterii edemului dintre celule.
Pe imagine: fibre musculare în distrofia musculară moderată progresivă:
a) fibre musculare violet deschis;
b) pete ușoare din interiorul fibrelor musculare – edem, împingând nucleii din centrul celulei spre periferie;
c) puncte întunecate - nuclee ale celulelor musculare;
d) săgeata arată o celulă musculară care nu se poate mișca din cauza scăderii proceselor metabolice – se întunecă spre violet.

- grad sever caracterizat prin focare extinse de distrugere a miofibrilelor, fragmentarea și dezorganizarea lor, apariția unei substanțe asemănătoare hialinei și edemîntre celulele musculare. Din punct de vedere funcțional, un astfel de țesut are o rezistență slabă, oboseala se instalează rapid și se dezvoltă semne de oboseală musculară. Fotografia va fi prezentată mai jos.
Aceasta este starea mușchilor din Emine înainte de a mă contacta:

Explicație pentru fotografie„grad sever de distrofie musculară”:
- Fibrele musculare din secțiune sunt colorate în albastru.
- Punctele roșii sunt nucleele celulelor musculare.
- Edemul este o culoare albă necolorată.
Tabloul clinic al distrofiei musculare
Primul semn al miopatiei Duchenne la Emine a fost slăbiciunea. A început să obosească de la efort fizic normal. Primele plângeri ale lui Emine au fost:
- Oboseală la alergare, mers lung.
- Emine a început să cadă des.
- Mialgia a început să apară la nivelul picioarelor (durere în mușchi), uneori în combinație cu spasme dureroase.
- Treptat, mersul a devenit dificil.
Emine nu se putea ridica de pe un scaun jos fără ajutorul mâinilor. Când s-a ridicat în picioare, femeia a recurs la utilizarea tehnicilor auxiliare: „stare în picioare cu o scară”, „cățărare singură” - tehnica Gowers. Câțiva ani mai târziu, Emine nu s-a putut ridica fără ajutor. Pacientul nu a putut să urce scările.
După impactul meu asupra mușchilor lui Emine, ea urcă la etajul 17 fără ajutorul mâinilor, coboară imediat liftul și urcă din nou la etajul 17 fără să se obosească!
Atrofia musculară se dezvoltă în principal în zona centurii pelvine, coapse (prin urmare, efectul emendic asupra mușchilor lui Emine a fost direcționat către aceste zone).
Mușchii extremităților superioare încep să se atrofieze mai târziu. Emine a spus că nu poate să-și toarne ceai sau să-și pieptene părul. Priviți rezultatele tratamentului distrofiei musculare în videoclipul de mai jos: